
Introduction: In sports, injuries are a very common occurrence, and managing them often requires a variety of treatments such as physiotherapy, surgery, bed rest, and medication. While not all sports-related injuries can be treated with medication alone, drugs play a crucial role in addressing certain symptoms. The primary types of medications used in sports include analgesics, anti-inflammatory drugs, certain antibiotics, and occasionally antipyretics like paracetamol.
Pain resulting from injuries, especially those common in contact sports, is typically managed with analgesics, which can be either narcotic or non-narcotic. Examples of narcotic analgesics include morphine, Damerol, and Dilaudid, while non-narcotic options include acetaminophen. Inflammation caused by injuries is addressed with NSAIDs (Non-Steroidal Anti-Inflammatory Drugs), with special emphasis on COX-2 inhibitors.
Setting aside general discussions, we will now focus specifically on the chemistry of these drugs—particularly the synthetic characteristics of analgesic and anti-inflammatory medications. This includes both widely marketed drugs and those still under research for their pharmacological potential in the immediate treatment of sports-related injuries.
Analgesics
Analgesics are defined as agents that alleviate pain by raising the pain threshold without affecting consciousness or altering other sensory modalities. From a psychological perspective, pain can be described as a specific type of sensory experience that the nervous system distinguishes from other sensations such as touch, heat, pressure, or cold. Pain is one of the most common, distressing, and significant symptoms associated with injury and many diseases. Rather than being viewed merely as a symptom, pain should be regarded as a syndrome of intensely unpleasant sensations. Pain is generally classified into two types: acute and chronic. Sports injuries typically result in acute pain. The following sections discuss analgesic and anti-inflammatory drugs commonly used in sports-related injuries.
Paracetamol (Acetaminophen: N-(4-hydroxyphenyl) acetamide)
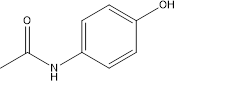
Figure 1
Synthesis:
- Catalyzed by a mineral acid, acetic anhydride reacts with the amino group of p-aminophenol. At first, the nucleophilic nitrogen atom of amino group of p-aminophenol attacks the carbonyl carbon of the acetic anhydride by nucleophilic addition mechanism (Fig.2). Then we get an intermediate compound which is further subjected to elimination of acetate anion, finally yielding the product acetaminophen (Fig.1) [2].
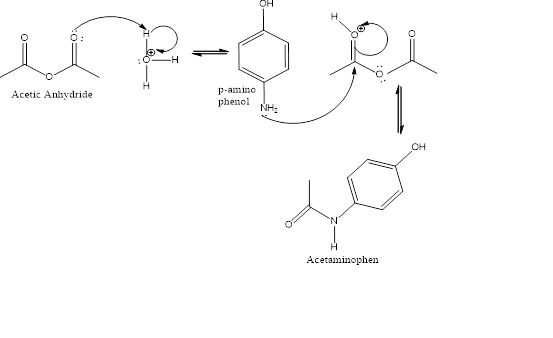
Figure 2
Further, the product may be re-crystallized using methanol, ethanol or propanol.
- An alternative industrial synthesis (Fig.3) developed by Hoechst–Celanese involves direct acylation of phenol with acetic anhydride catalysed by HF, conversion of the ketone to a ketoxime with hydroxylamine, followed by the acid-catalysed Beckmann rearrangement to give the amide[3]
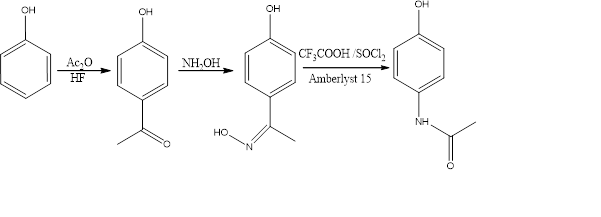
Figure 3
Therapeutic uses: It has analgesic activities, but relatively less anti-inflammatory activity. Paracetamol is popularly used to reduce mild to moderate pain and occasionally severe pain. [4]. Paracetamol is available globally in large scale including in India in different brand names in tablet form.
- Aspirin (Acetylsalicylic acid)
Acetylsalicylic acid or Aspirin (Fig.4) was first synthesized by Gerhardt in 1852, but unfortunately this wonder drug, more or less remained obscure until Felix Hoffmann studied its detailed pharmacodynamic properties in 1899. It gained entry into the world of medicine through Dreser, who coined a new name ‘aspirin’ derived from “a” of acetyl and adding to it “spirin”, an old name of salicylic or spiric acid, obtained from spirea plants [5].
Salicin was the first compound belonging to salicylic acid derivatives category that exhibited medicinal value. It was employed as a substitute for quinine as a febrifuge. In 1838, Paria prepared salicyclic acid and whose structure was established by Hoffmann. Kolbe and Lautermann, (1860) introduced the commercial method of preparing salicyclic acid from sodium phenate.
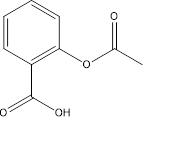
Figure 4
Synthesis:
- Ethanoic anhydride is used to convert 2-hydroxybenzoic acid into aspirin (2- acetoxy benzoic acid). The reaction takes place easily in acidic solution but the product is formed as part of a mixture containing several other compounds.
- A two-step reaction involves converting oil of wintergreen – natural product, into salicylic acid which is further converted into aspirin. The steps involved are deprotonation, attack by hydroxide ion using sodium hydroxide solution, removal of methanol and protonation of dianion. The reaction mixture needs to be boiled and cooled to finally obtain crystals [6]. The crystals of salicylic acid are boiled in a reaction mixture along with acetic anhydride and phosphoric acid as a catalytic reagent. The crystals of aspirin are formed on cooling in room temperature.
- A very innovative and greener way of aspirin synthesis is by using microwave irradiation [7], using salicylic acid and acetic anhydride (Fig. 5). The research- group reported 80% yield without using any catalyst. It was performed completely without any use of solvent, thus solid-state reaction. AlCl3, CaCO3, NaOAc, Et3N and DMAP were used as catalysts and gave positive results in microwave heating. Re-crystallisation was done by using toluene as solvent.

Figure 5
Therapeutic uses: Aspirin helps to reduce pain as well as inflammation. Hence it can also be called an anti-inflammatory drug. It is quite readily available in market and in India it is sold under different brand names.
3. Salicylamide:
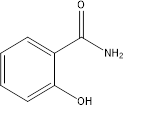
Figure 6
Synthesis:
- Salicylamide (Fig.6) can be synthesised easily from urea and phenol as primary reagents (Fig. 7), however without any catalyst the yield was very poor. On using of certain Mg/Al catalyst the yield was found to increase drastically [8]. MgO, ZnO, MgAlO-2, MgAlO-3, MgAlO-4 catalysts were tested which gave yield of salicylamide as 34%, 45%, 53.5%, 45.7%, 31.3% respectively.
Figure 7
- Another efficient means to synthesise salicylamide derivative (Fig.8) was reported by Lei Zhang and John Y. Zhang (2006). They proposed the BCl3 mediated coupling reaction between phenol and isocyanate to synthesise salicylamide scaffolds [9]. Boron trichloride specially imparts an orthoselectivity to the ring. They used microwave irradiation to speed up the process. Higher temperature proved to increase the yield as well as speed up the process of conversion. For the preparation of 2-hydroxy-N-(p-tolyl) benzamide, 140˚C for 10 mins proved to be the most efficient.
- A general lab preparation of salicylamide is reported by making salicyl chloride obtained by treating salicylic acid with PCl5, and then aqueous ammonia is added till the completion of the reaction.

Figure 8
Therapeutic activity: Its analgesic activities are similar to that of aspirin. It may be used in place of salicylates where apparent sensitivity occurs. It is widely used as an ingredient along with acetylsalicylic acid, acetaminophen, paracetamol etc. in several analgesic, pain relieving drugs in India.
4. Diflunisal:

Figure 9
Synthesis:
Diflunisal (2’,4’-Difluoro-4-hydroxy-[1,1’-biphenyl]-3-carboxylic acid: Fig.9) has been prepared with different synthetic routes which can be divided into conventional methods and catalytic methods. Three different schemes for the synthesis of diflunisal are as follows: –
- Five step synthesis of Diflunisal by Hannah et al, (Fig.10) consisted of a Benzene arylation using a Gomberg-Bachman-Hay reaction, Friedel craft’s acylation, Bayer Veliger oxidation, saponification and carbonation of phenol via Kolbe Schmidt reaction with the yield about 35.78% [10].
Reaction Conditions for figure 10:- (I)-Benzene, i-amylnitrile,67˚C 1,5h reflux 2h, 25˚C, 2 days.; (II)- Acetic anhydride,CS2,AlCl3,Reflux,30 min.; (III)- a) trifluro acetic anhydride,DCM,H2O2,, b) DCM,Na2HPO4, reflux,1h.; (IV)- NaOH,water reflux 2h.; (v)- CO2, K2CO3,3400psi 230˚C,6h.
Figure 10
- Preparation of Diflunisal via two steps by Giordano et al (Fig.11): In this process phenylboronic acid was made from the corresponding Grignard reagent (gave a 83.4% yield). Pd(PPh3)4 was used as a catalyst for the Suzuki reaction giving a 59% Diflunisal yield.
Reaction conditions for figure 11: – (I) a) THF, B(OMe)3,N3, -15˚C,1h.; b) H2SO4,0˚C,30 mins.; (II) Pd(PPh3)4,Na2CO3,ethanol,80˚C,3h.
Figure 11
- Preparation of Diflunisal by Suzuki reaction in only one step (Fig.12) using Phosphine based catalyst, glovebox and degassed water [11] was also reported with yield of 95%.
Figure 12
Therapeutic activity: It is a strong analgesic used to cure musculoskeletal sprain, injuries to tendons as well as inflammation and strains from minor sports injury. Its analgesic effect is longer than that of aspirin. It was first introduced in market by Merc & Co. under different brand name. It is widely available in the market.
5. Celecoxib and structural analogues:
Celecoxib is an effective NSAID, selective COX 2 inhibitor, having a heterocycle as the main structural feature (Fig. 13). It has three modification sites in its structure which can be modified to bring about small changes to its activity with the aim to make it more potent and reducing risks of digestive problems including ulceration etc.
Figure 13
Synthesis:
- By 1,3 dipolar cycloaddition reaction:Reaction of benzene sulphonates with enamines precursors resulted in the isolation of the pyrazole containing celecoxib drug with good regioselectivity [12].
- By coupling reaction: Organ and coworkers reported the synthesis of 4-(5-iodo-3-methylpyrazolyl)phenylsulfonamide and relevant coupling analogs [13].It involves a 6-step reaction with a Pd catalyst.
- Amgad G. Habeeb, P. N. Praveen Rao, and Edward E. Knaus[14]in 2001 had developed a scheme to produce a azido analogue of both Rofecoxib and celecoxib. The SO2NH2 here has been replaced by a N3 group. The new analogue has shown similar analgesic properties and is found to produce COX-2 inhibition.
4-Nitrophenylhydrazine and 1-(4-methylphenyl)-4,4,4-trifluorobutane-1,3-dione were used as the initial reactants. A series of 4 steps are involved in the synthesis (scheme 1). NO2 group was reduced to amino group by hydrazine hydrate and 10% Pd/C. Then diazotisation was performed in the conventional way to yield the final celecoxib azido analogue.
4-Azidoacetophenone was used as the precursor for the rofecoxib analogue. It was brominated by Br2. The substrate was then esterified with phenyl acetic acid and finally after a reaction with Et3N the final product, final product could be obtained. Reaction conditions for, Scheme 2: (I)-EtOH, HCl, reflux, 20 h; (II)- H2NNH2‚xH2O, 10% Pd/C, reflux, 45 min; (II)- NaNO2/HCl and then NaN3, 0-5 °C, 45 min,; Scheme 2: (I) Br2, CHCl3, 25 °C, 2 h; (II) PhCH2COOH, Et3N, CH3CN, 25 °C, 1 h; (III) Et3N, CH3CN, reflux, 8 h.
Scheme 1
Scheme 2
6. 1,5-Diarylpyrrole-3-acetic acid and ester derivatives:
A) Biava and co-workers in 2005 synthesised (Fig. 14) some new pyrrole containing anti-inflammatory drugs [15]. They have synthesised 1,5-diarylpyrrole-3-acetic acids and esters which have shown considerable COX-2 inhibitory activities. It contains a pyrrole acetic and vicinal diaryl heterocyclic moiety. The insertion of the acetic moiety in this coxib analogue gave rise to very potent and selective COX-2 inhibitor. The penultimate compound in the synthetic path which was an ester derivative of the acid with the alkyl R group as CF3 and H had potent COX-2 inhibitory activity. Also, the final acid derivative having the R group as CF3 also showed potent COX-2 inhibitory activities.
Synthesis:
4-(Methylthio)benzaldehyde with methyl vinyl ketone is the main starting reagent for the initial synthesis of 1,4-diketone. This was converted into corresponding methyl sulphonyl derivative by means of an Oxone oxidation. This compound was cyclised by Paal-Knoor condensation to yield the expected 3-unsubstituted 1,5-diarylpyrrole. Then the acetic acid group was incorporated by a regioselective acylation with ethoxalyl chloride in presence of pyridine. This keto ester was reduced by triethylsilane to give pyrrole acetic esters which were hydrolysed by KOH to get the expected acids.
Figure 14
Reagents and conditions: (I) CH2CHCOCH3, NEt3, EtOH, 75-80 °C, 20 h, thiazolium; (II) Oxone, MeOH/H2O, room temp, 2 h; (III)RPhNH2, TiCl4, toluene, room temp, 10 h; (IV) EtOCOCOCl, Pyridine, CH2Cl2, 0 °C, 4 h; (V) Et3SiH, CF3COOH, room temp, 24 h; (VI) KOH, EtOH, reflux, 2 h.
B) In 2010 Biava and coworkers performed a modification in the previously made analogue in 2005[16]. They simply introduced a long side chain of ‘CH2COOR’ in the C3 position of the pyrole ring. This ultimately proved to be a better substrate for COX-2 inhibition, showing very high selectivity index.
Synthesis: –
The initial substrate is taken as 4-(methylthio)benzaldehyde and methyl vinyl ketone. The reaction was done in a microwave apparatus. The 4-methyl sulfonyl derivative was made by oxone oxidation of the intermediate formed in the previous step. Next step is the Paal-Knoor condensation with the desired arylamine in a microwave apparatus for 45 mins. It was then cyclised to achieve the diaryl pyrrole. then the C3 side chain was formed by regioselective acylation with oxalyl chloride. Then esterification was done by reaction with appropriate alchohol. Finally, the alpha-keto ester derivatives were reducved by triethylsilane in trifluoracetic acid to get the desired pyrole esters.
In figure 15 compound A of the target molecule shows maximum selectivity index of >13600 for COX-2. Compound B, E, G, D, F, C all has selectivity index above >2300 arranged in decreasing value. In the second step X=SMe was converted to SO2Me similarly as shown in Figure 15.
Reaction conditions for figure 15: – (I)CH2dCHCOMe, TEA, 3-ethyl-
5-(2-hydroxyethyl)-4-methylthiazolium bromide, MW, 15 min; (ii) Oxone, MeOH/H2O, room temperature, 2 h; (II) RPhNH2, p-toluenesulfonic acid,EtOH, MW, 45 min;
(III) ClCOCOCl, 2,6-lutidine, CH2Cl2, R1OH; (IV) Et3SiH, TFA, room temperature, 2 h.
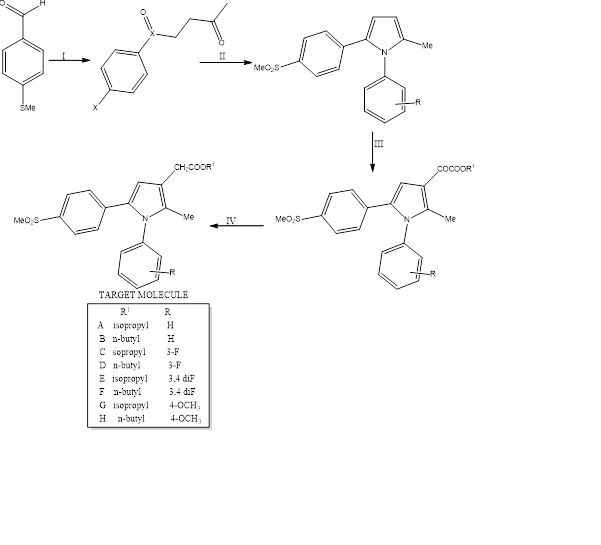
Figure 15
7. 1,5-Diarylpyrrole-3-Alkoxyethyl ester derivatives:
Initially these derivatives have been created which show good COX-2 inhibitory activities [17]. Replacement of the acetic ester group with an alkoxy group showed a considerable increase in COX-2 inhibitory activity. Anzini and co-workers in 2008 has replaced the ester moiety with three different alkoxy group namely methyl, ethyland n-propyl group. Some of the derivatives have a considerable anti-inflammatory and antinociceptive activities.
Synthesis:
Anzini and co-workers synthesised the 1,5- diaryl-3-(2-alkoxyethyl) pyrroles starting from an appropriate ethyl acetate ester (Fig. 16) which was previously prepared and had inhibitory activities [16]. Using LAH as reducing agent in dry THF at room temperature the ester moiety was converted to its corresponding alcohol. Then it was reacted with the required alkyl iodide in presence of powdered KOH in DMSO which yielded the corresponding alkoxide in considerable yield.
Figure 16
Reaction conditions: (I)- LiAlH4, THF, room temp,20 mins; (II)- KOH, RI, DMSO, room temp,30 mins.; where R = Me, Et, n-Pr; X = H, CF3.
9. Water-Soluble Sulfonylphosphoramidic Acid Derivatives of the COX-2 Selective Inhibitor Cimicoxib :
In the case of intense pain due to severe injuries, immediate action of analgesics are desired and so parenteral treatments are followedfor rapid onset of actions. In these context drugs are preferred to be water soluble in nature for very obvious reasons. Most potent market available COX-2 inhibitors are celecoxib, rofecoxib, valdecoxib. Almansa and co-workers in 2004[18]have prepared a water soluble phosphonamidite derivative of cimicoxib (Fig. 17), which itself is a good COX-2 inhibitory drug but not water soluble. Due to the presence of the NH group it can form salts and hence become water soluble.
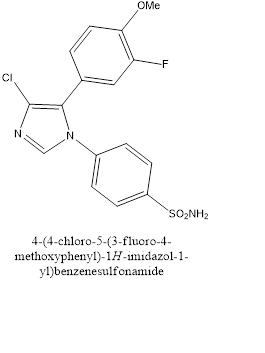
Figure 17
Synthesis:
The initial cimicoxib compound is the starting compound. Corresponding dialkyl chlorophosphite (ClPO(OR)2), NaOH is used to convert it to the phosphoramidate. The OR group attached to the P is converted to ONa using (NaI in acetone; Finkelstien’s reaction). Secondly the phosphamidate is converted to phospharimidic acid by using iodotrimethylsilane. The sodium and potassium salts were made by NaOH and KOH and then recrystallised. Reaction steps (Figure 18): (I)-2N NaOH,CIPO(OR)2,THF, 20˚C,6h; (III)-NaI,acetone,50˚C,18h; (II)- (1) ISiMe3,DCM,0˚C,I h, (2) H2O/acetone,20˚C,18h
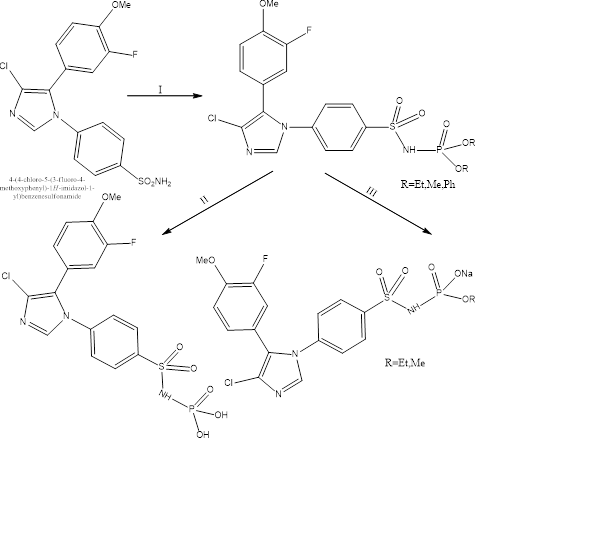
Figure. 18
References:
(1) Friderichs, E.; Christoph, T.;Buschmann, H.; Ullmann’s Encyclopedia of Industrial Chemistry, 2011, doi:10.1002/14356007.a02_269.pub2.
(2) Srabovic, M.; Huremovic M.; Catovic, B.; Kulic S.; Taletovic A.; J. Chem. Bio. Phy. Sci. (Sec. A), 2017, 7(1), 218-230.
(3) US patent 4524217 by Kenneth, G. D.; Charles, B. H., (assigned to Celanese Corporation);The American Society of Health-System Pharmacists, Archived from the original (1985) on 5 June 2016 and Retrieved on 16 September 2016.
(4) Gringauz, Alex; Introduction to Medicinal Chemistry. 1st edition, Wiley-VCH, 1996.
(5) Kar, A., Medicinal Chemistry,4th edition, New Age International(P) Limited Publishers, India, 2007, 280-282.
(6) Olmsted, J. A., Journal of Chemical Education,1998, 75(10), 1261-1272.
(7) Montes, I.; Sanabria, D.; García, M.; Castro, J.; Fajardo, J., Journal of Chemical Education,2006,83(4), 628-634.
(8) Wang, D.; Zhang, X.; Wei, W.; Sun, Y.,Catalysis Communications,2012, 28, 159–162.
(9) Zhang, L.; Zhang, J. Y., Journal of Combinatorial Chemistry,2005,7(4), 622–626.
(10) Kylmälä, T.; Tois, J.; Xu, Y.; Franzén, R., J. Open Chemistry,2009, 7(4), 68-73.
(11) Kylmälä, T.; Tois, J.; Xu, Y.; Franzén, R., J. Open Chemistry, 2009, 7(4), 478-484.
(12) Hamme, A.; Dadiboyenal, S., Current Organic Chemistry,2012. Vol-16, Bentham Science Publishers,USA.
13) Organ, M. G.; Mayer, S., J. Comb. Chem.,2003, 5, 118-124.
(14) Habeeb, G.; Rao, P. N. P.; Knaus, E. E., J Med Chem, 2001, 44(18), 3039-3042.
(15) Biava, M; Porretta, G, C; Cappelli, A; Vomero, S; Manetti, F; Botta, M; Sautebin, L; Rossi, A; Makovec, F; Anzini, M; J. Med. Chem., 2005., 48(9), 3428-3432.
(16) Biava, M.; Porretta G. C.; Pose, G.; Battilocchio, C.; Manetti, F.; Botta, M.; Forli, S,; Sautebin, L,; Rossi, A,; Pergola, C.; Ghelardini, C.; Galeotti, N.; Makovec, F.; Giordani, A,; Anzellotti, P.; Patrighnani, P,; Anzini, M., J. Med. Chem., 2010, 53, 1269-1276.
(17) Anzini, M; Rovini ,M; Cappelli, A; Vomero, S; Manetti, F; Botta, M; Sautebin, L; Rossi, A; Pergola, C; Ghelardini, C; Norcini, M; Giordani, A; Makovec, F; Anzellotti, P; Patrignani P; Biava, M;. J Med Chem 2008, 51(15), 4476-4481.
(18) Almansa, C; Bartrolí, J; Belloc, J; Cavalcanti, FL; Ferrando, R; Gómez, LA; Ramis, I; Carceller, E; Merlos, M; García-Rafanell, J.; J. Med. Chem., 2004.47(22), 5579-5582.