
Introduction:
Oxaliplatin (OXA) is a third-generation platinum-based antitumor agent with a broad spectrum of anticancer activity[^1]. Structurally, it is a pseudo-symmetric dicarboxylic acid that is less toxic, and its crystal structure corresponds to the space group P2₁/m[^2]. Unlike cisplatin and certain other platinum drugs such as carboplatin, OXA does not exhibit cross-resistance[^3]. It is widely used in chemotherapy for tumors of the digestive system, including gastric, colorectal, and pancreatic cancers[^4]. Although it is considered a potential carcinogen, the International Agency for Research on Cancer has not yet officially classified it as such. Following intravenous administration, OXA is primarily excreted through the urine, which is the typical route of elimination in humans[^5].
Oxaliplatin-induced peripheral neuropathy (OIPN) presents in two main forms: acute and chronic. Acute OIPN generally appears within hours of intravenous infusion and is characterized by symptoms such as peripheral neuritis in the limbs or mouth, sensory disturbances, and paresthesia. Patients may also experience altered tongue sensation and speech difficulties, though these effects are usually short-lived[^6]. Chronic OIPN, on the other hand, is among the most frequent adverse effects caused by OXA and other chemotherapeutic agents. Its clinical manifestations include persistent paresthesia, limb numbness, spasms in the upper respiratory tract and perioral muscles, allodynia, hyperalgesia, and sensory loss that may impair daily activities.
The mechanisms underlying acute OIPN involve changes in transient receptor potential channels, voltage-gated sodium, potassium, and calcium channels, as well as the OCT2 transporter protein and glial cells. Chronic OIPN is associated with DNA damage, mitochondrial dysfunction, oxidative stress, glial activation, and neuroinflammation[^7–12].
Chronic neuropathy tends to be long-lasting and can persist even after cessation of treatment. As chemotherapy progresses and the cumulative dose of OXA increases, symptoms such as limb numbness, paresthesia, and sensory pain become more pronounced[^13]. When cumulative doses reach 650–700 mg/m², approximately 10% of patients experience persistent neuropathic symptoms. At doses of 780–850 mg/m², the incidence rises to around 30%, and when the total cumulative dose exceeds 1560 mg/m², nearly 75% of patients are affected. Typically, after 4–6 chemotherapy cycles, the cumulative dose ranges from 520 to 780 mg/m²[^14].
Peripheral neuropathy is the most prevalent side effect of OXA treatment. About 50% of patients receiving OXA over a 5–7 month period develop moderate to severe neuropathy, in a dose-dependent and cumulative manner, which can limit both dosing and therapeutic effectiveness[^15]. This review aims to explore the long-term consequences, risk factors, underlying mechanisms, and available treatments for chronic OIPN.
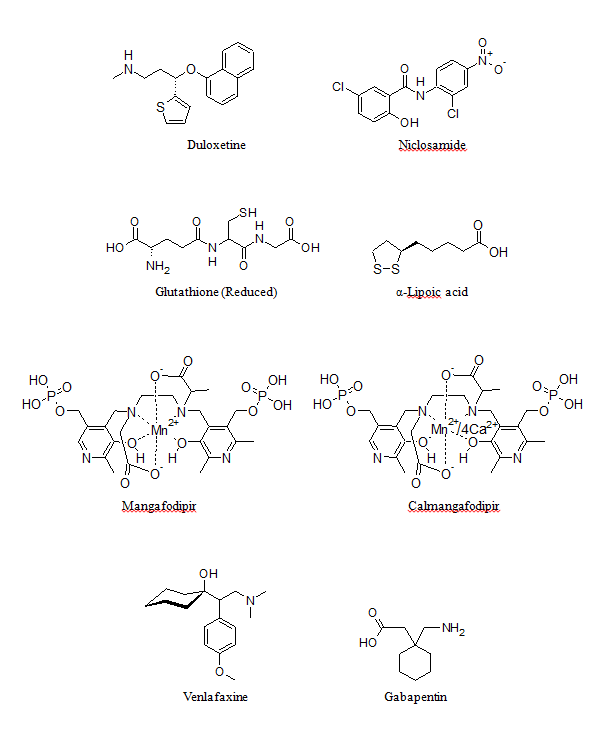
Figure 1: Structures of agents used for the management of OIPN.
2. Risk factors associated with OXA usage:
2.1. Treatment plan:
Risks associated with the treatment plan typically involve factors such as dosage, injection timing, treatment duration, and monitoring for peripheral neuropathy during each chemotherapy cycle[^16]. A systematic review assessed the neurotoxic effects persisting 12 months or more after the cessation of oxaliplatin (OXA) treatment. This review encompassed 14 studies, including 3,869 colorectal cancer (CRC) patients. The majority had received OXA combined with fluorouracil-based chemotherapy regimens, with total OXA doses ranging from approximately 680 to 1450 mg/m² and median dose intensities between 30.8 and 42.6 mg/m²/week. Most studies used the Common Terminology Criteria for Adverse Events to assess neurotoxicity, which ranged from grades 0 to 3. Of the six studies that directly analyzed the correlation between neurotoxicity and cumulative OXA dose, five confirmed a significant association, while one study did not observe this relationship. The review concluded that a considerable number of patients continued to experience neurotoxic symptoms even 12 months after discontinuing OXA. Moreover, it was noted that higher cumulative doses of OXA could contribute to long-term neurotoxicity[^17].
The two common chemotherapy protocols incorporating OXA—FOLFOX6 (OXA with 5-fluorouracil/leucovorin) and XELOX (OXA with capecitabine)—show varying levels of neurotoxicity. A comparative study examining these regimens in the primary treatment of metastatic colon cancer found no significant difference in the overall incidence of oxaliplatin-induced peripheral neuropathy (OIPN) using NCI-CTC assessment criteria. The incidence was 90% for the XELOX group (155 patients) and 95% for the FOLFOX6 group (149 patients). However, severe neurotoxicity (grades 3/4) was notably less frequent in the XELOX group (11%) compared to the FOLFOX6 group (26%)[^18]. A head-to-head clinical trial involving 150 CRC patients treated with either XELOX or FOLFOX4 further evaluated neurotoxicity using NIP-CTC version 3, clinical total neuropathy scores (TNSc), and electrophysiological assessments. While the incidence and severity of acute neurotoxicity were similar between groups, FOLFOX4 was more strongly associated with chronic neurotoxicity at comparable cumulative OXA levels (64/77 vs. 44/73; p = 0.002)[^19].
Another clinical study involving 106 CRC patients who had undergone at least two lines of OXA-based therapy explored whether subsequent OXA treatment exacerbates existing neuropathy or causes new symptoms. After approximately 30 months and an average of eight OXA cycles, a significant proportion of patients developed grade 1 (about 40%) and grade 2 (around 23%) OIPN, while no cases of grade 3 were observed. About one-third (32%) of participants showed worsening of existing OIPN symptoms. The study concluded that cumulative OXA dose was independently associated with increased OIPN risk. Nevertheless, re-treatment with OXA was considered a viable option even for patients with prior mild to moderate OIPN[^20].
2.2 Genetic polymorphisms related to OIPN:
2.2.1. Voltage-gated channel genetic polymorphisms:
At this juncture, most of the study focuses on OIPN voltage-gated Na+ channel genetic polymorphism, but there are controversies that remain. For instance, one such study performed by Argyriou et al., 2009, genotyped 62 patients with advanced colon cancer who received chemotherapy with OXA, and studied the relationship between OIPN and SCN2A R19K gene polymorphism. 36 cases (58.1%) produced OIPN, 14 cases (38.9%) had grade 1 neurotoxicity, 22 cases (61.1%) had grade 2 neurotoxicity. Adding to this, the proportion of homozygous and non-homozygous OIPN was similar. It was concluded that there was no obvious causal relationship between SCN2A R19K gene polymorphism and OIPN 21. Another study conducted by the same research group yet again with a sample of 200 patients, concluded that the relationship appeared close: 72.5% of patients (145/200) developed cumulative OIPN after discontinuation. Among them, heterozygous rs2302237 is suggestively connected to the prevalence of chronic OIPN (OR: 2.47, p = 0.037) 22.
A clinical trial was used to determine the possibility of variations in SCNA gene being linked to the enhanced vulnerability of developing chronic OIPN in patients with cancers of the GI tract experiencing OXA treatment. More than 200 GI tract cancer patients, who received OXA in order to suppress secondary or metastasized tumors or to shrink a tumor before the intended main therapy were enrolled for the study. Genotyping was performed by real-time polymerase chain reaction (RT-PCR). It was established that rs6746030 as a chromosomal variant of SCN9A was pointedly concomitant with a greater occurrence of chronic OIPN (OR=1.8), while, the rs6754031 variant was related to a less significant occurrence (OR=0.45). It was also interesting to note that the SCN10A variant was linked to increased severity of chronic OIPN (OR=2.0). The study provided an indication that a pivotal relationship exists concerning chronic OIPN and voltage-gated sodium channel polymorphisms 23.
2.2.2. Glutathione S transferase genetic polymorphism:
Glutathione S transferase is currently the most studied gene related to OIPN genetic polymorphism. It is divided into at least 5 subtypes, among which GSTP1 is highly expressed in colon cancer and participates in the detoxification process of platinum drugs. It may also be related to the resistance developed for platinum based drugs 24. Several studies have shown a relationship between GSTP1 genetic polymorphism and OIPN. However, a systematic review of the relationship between GSTP1Ile105val polymorphism and OIPN conducted by Peng et al., 2013, concluded that there was no direct correlation between the two phenomena 25. Therefore, the correlation between GSTP1 gene polymorphism and OIPN still remains controversial.
2.2.3. Whole genome polymorphism analysis:
Won et al. (2012) conducted a prospective genome-wide study involving colon cancer patients undergoing oxaliplatin (OXA)-based chemotherapy, aiming to identify potential genetic markers linked to chronic oxaliplatin-induced peripheral neuropathy (OIPN). Peripheral blood DNA was extracted from patients, and the primary endpoint was defined as the occurrence of severe OIPN—either Grade 2 neurotoxicity lasting more than seven days or Grade 3 neurotoxicity lasting fewer than seven days. Ninety-six patients receiving adjuvant chemotherapy with OXA plus fluorouracil were genotyped using a human genome-wide SNP array. An additional 247 patients with advanced disease treated with OXA were included for validation. The results identified several gene polymorphisms significantly associated with chronic OIPN, including TAC1 (Tachykinin), FOXC1 (Forkhead box C1), ITGA1 (Integrin alpha 1), CAMK2N1 (calcium/calmodulin-dependent protein kinase II inhibitor 1), BTG4 (B-cell translocation gene 4), and FARS2 (phenylalanyl-tRNA synthetase 2). These findings suggest that screening for these genes may help predict the likelihood of developing chronic OIPN.
In a separate study, Terrazzino et al. (2015) assessed 150 Caucasian colorectal cancer (CRC) patients receiving OXA-based chemotherapy to evaluate the impact of eight specific genetic variants (rs10486003, rs2338, rs843748, rs797519, rs4936453, rs12023000, rs17140129, and rs6924717) previously implicated in genome-wide association studies as potential predictors of chronic OIPN. According to the NCI-CTC criteria, none of the polymorphisms showed a significant association with grade ≥2 chronic OIPN. However, a nominal association was observed for ACYP2 rs843748 using the TNSc scale (dominant model: OR 0.27, 95% CI: 0.10–0.75, P = 0.008). The authors proposed that these SNPs may still hold relevance and warrant further investigation as possible genetic determinants of chronic OIPN.
A large-scale study was also undertaken to explore genetic predictors of response and tolerance to adjuvant-modified FOLFOX6 (mFOLFOX6) in over 850 patients with stage II or III colon cancer post-surgery. Of these, 465 patients were suitable for evaluating the influence of individual genotypes on the onset of OIPN. When comparing patients with grade 0/1 versus those with grade 2/3 or grade 3 OIPN, no significant associations were found for any of the 12 SNP markers, even when accounting for total OXA dose. The authors concluded that none of the markers showed a meaningful connection to the development of OIPN.
Moreover, other studies have pointed to potential roles for additional genetic factors, such as AGXT, genes encoding various proteins, and base excision repair polymorphisms (ERCC1, XRCC1), as well as previously mentioned genes like FARS2, ACYP2, and TAC1, in influencing patient susceptibility to OIPN. Besides genetic predispositions, clinical factors such as a history of diabetes, older age, severe anemia, hypoproteinemia, hypomagnesemia, and alcohol consumption also appear to contribute to the risk of developing OIPN.
3. Neuropathies induced by OXA usage:
3.1. Dorsal root ganglion (DRG) injury hypothesis:
The dorsal root ganglion (DRG) is a primary sensory neuron. In neuropathic pain models resulting from various causes, the key electrophysiological alteration in the DRG is the subthreshold membrane potential oscillation in neurons. This leads to ectopic spontaneous discharges, which in turn cause abnormal behaviors in animals, such as spontaneous pain, heightened sensitivity to pain (hyperalgesia), and pain triggered by touch. These responses can be regarded as indicators of chronic pain.
3.1.1. Platinum drug accumulation in DRG:
Multiple studies have identified the dorsal root ganglion (DRG) as a primary site of chronic neurotoxicity caused by platinum-based compounds. Oxaliplatin (OXA) induces the production of reactive oxygen species (ROS), such as superoxide anions and hydroxyl radicals, which can result in DNA strand breaks within cells. The DRG is particularly vulnerable because it lacks the protection of the blood-brain barrier and has a slow metabolic rate, limited detoxification capacity, and reduced DNA repair efficiency in response to platinum-induced damage[^34].
A study was conducted to evaluate the impact of OXA on cell viability, DNA damage, ROS generation, and functional parameters in cultured rat sensory neurons, both with and without reduced expression of the base excision repair (BER) protein AP endonuclease/redox factor-1 (APE1). The results showed that OXA aggravated genomic damage, as evidenced by increased levels of phospho-H2AX and a reduction in capsaicin-stimulated release of calcitonin gene-related peptide (CGRP) from the neurons[^35].
In a separate experiment, McKeage et al. (2001) reported that a single dose of OXA (10 mg/kg) did not significantly affect the size of DRG neuronal cell bodies or nuclei within several hours post-administration. However, a marked reduction in nucleolar size was observed, peaking at 24 hours and recovering slowly. This reduction showed a nonlinear correlation with the OXA dose (r² = 0.99). Functional neurotoxicity became evident 14 days after treatment, with recovery noted after three months. During repeated dosing, nucleolar size changes were linearly associated with the progressive alteration in sensory nerve conduction velocity (p < 0.024). These findings suggest that chronic oxaliplatin-induced peripheral neuropathy (OIPN) may result from repeated nucleolar damage in sensory neurons, which inhibits rRNA synthesis, disrupts protein production, and ultimately causes morphological abnormalities and functional impairment of neuronal organelles[^36].
3.1.2. pNF-H deletion:
The phosphorylated neurofilament heavy subunit (pNF-H) plays a key role in neurofilament formation and offers protection to neuronal cells against damage. According to Jamieson et al. (2009), treatment with OXA led to a reduction in pNF-H immunoreactivity in neuronal cells, although no such change was observed in the L5 DRG nerve fibers. Morphological assessments revealed significant reductions in both the number (75%; p < 0.0002) and size (45%; p < 0.0001) of pNF-H-immunoreactive neurons following OXA administration. When comparing the effects of various chemotherapeutic agents, a notable decrease in the number of pNF-H-positive neurons was found with OXA (77.2%; p < 0.0001) and cisplatin (35.2%; p = 0.03), but not with carboplatin or paclitaxel. Additionally, a significant reduction in the average cell area of pNF-H-labeled neurons was observed only after OXA treatment (31.1%; p = 0.008), with no significant changes noted after cisplatin, carboplatin, or paclitaxel administration. These findings suggest that the development of OIPN is more closely associated with the reduction of pNF-H in rat DRG tissues.
3.1.3. MAPKs family includes three subfamilies:
The first three subfamilies are extracellular-signal regulated protein kinase (ERK), c-Jun N-terminal protein kinase (JNK) and p38 mitogen-activated protein kinase (p38). Among them, ERK1/2, JNK/Sapk and P38 play an important role in many peripheral neuropathies including chemotherapy-induced neuropathy. Long-term exposure of DRG neurons to OXA induces early activation of p38 and ERK1/2, which mediates neuronal apoptosis, while drug therapy can down-regulate JNK/Sapk for neuroprotection 38.
3.1.4. NR2B protein:
This subunit protein, typically produced by the DRG cluster at the beginning, gradually distributes into the supersynaptic membrane of the superficial dorsal horn in the spinal cord. The N-methyl-D-aspartate receptor subtype 2B (NR2B) of this region binds to glutamate, activating the N-methyl-D-aspartate (NMDA) receptor, which increases Ca²⁺ permeability and causes the breakdown of intracellular neurofibrin, ultimately resulting in nerve cell death. Consequently, inhibiting the NR2B subunit provides neuroprotective benefits. Mihara et al. (2011) demonstrated that mechanical allodynia developed by the fourth week after repeated administration of OXA (4 mg/kg, intraperitoneally, twice weekly), which was later reversed by intrathecal injection of the NMDA receptor antagonist MK-801 (10 nmol/L) and dimethylammonium (1 μmol). Likewise, the use of NR2B antagonists Ro25-6981 (300 nmol, intrathecally) and Effendil (50 mg/kg, orally) significantly reduced the OXA-induced pain behavior. Furthermore, elevated expression of NR2B protein and mRNA in the rat spinal cord was observed 25 days (late phase) after OXA treatment, rather than at 5 days
3.1.5. Changes in TRPs channels:
Transient Receptor Potential (TRP) superfamily consists of non-selective cation channel proteins that are widely present in cell membranes or intracellular organelle membranes. The TRP family consists mainly of seven subfamilies. In vivo, the specific cellular signaling pathways, including PAR2 and downstream enzymes PLC, PKC and PKA, increased TRPV1, TRPA1 and TRPV4 sensitivity in chemotherapy-related chronic neurotoxicity 41. Ta et al., 2010, used RT-PCR to analyze in vitro and in vivo TRPV1, TRPM8 and TRPA1 expressions induced by OXA. In vitro, embryonic rat DRG neurons were cultured for 48 hours using OXA. The expression of TRPV1, TRPM8 and TRPA1 mRNA in DRG neurons increased significantly after platinum, and TRPV1 may play a critical role in cisplatin-induced thermal hyperalgesia in vivo 42. Gauchan et al., 2009, studied whether TRPM8 (activated by hypothermia) was involved in the pain mechanism induced by cold stimulation of OXA. The results confirmed that OXA could induce the up-regulation in the expressions of TRPM8 mRNA and proteins in rat DRG cells 43.
3.2. Axonal neuropathy:
Repeated cumulative exposure to OXA and long-term increase in the excitability of Na+ channels can interfere with axonal ion conduction and cause damage to nerve cells. A clinical study evaluated the nerve conduction studies and neuronal excitability in 16 patients treated with OXA. 50% of patients had symptoms confirmed by these studies, which included abnormal nerve conduction, low sensory potential, and normal motor nerves. After 12 months of follow-up in symptomatic patients, the symptoms of peripheral neurotoxicity improved, but abnormalities in nerve conduction still existed. The threshold of axons was high and the nerve excitability was more significant. The refractory nature of this group of patients was more obvious (symptomatic group: 56.3 +/- 24.9%; whole group: 46.3 +/- 12.5%; control group: 27.1 +/- 1.9%; p<0.05) 44.
4. Apoptosis and other toxicity mechanisms for OIPN:
Donzelli et al., 2004, studied the effect of OXA on human neuroblastoma SH-SY5Y cells. The optical density analysis showed that the amount of proapoptotic protein p53 was significantly increased after exposure to OXA, and the total expression of anti-apoptotic protein Bcl-2 was significantly reduced. Caspase 3 and 7 are activated, but at a later stage, suggesting an auxiliary role in the process of apoptosis. Among the mitogen-activated protein kinases, only the p38 protein is activated early (by phosphorylation), which may play a notable role in inducing apoptosis. Therefore, it has been concluded that the occurrence of OIPN is associated with specific molecules involved in regulating the balance between the cell cycle and apoptosis 45. A literature-based review describes the mitochondrial mechanism underlying OIPN and the neuropathic complications associated with different antineoplastic agents. It shows that various neuropathic disorders as well as OIPN are due to mitochondrial impairment, relevant impairment of Ca2+ signalling pathways and reactive oxygen species (ROS) related oxidative stress that ultimately leads to apoptosis 46.
Oxidative stress is a key factor in the induction of OXA-associated peripheral neuropathies 47-49. This oxidative stress has also been linked to transient receptor potential cation channel TRPA1, which acts as a sensor for electrophilic and reactive species generated during tissue injury and inflammation 50. It is thought that by-products generated by ROS following OXA treatment could activate TRP1A, thereby producing a nociceptive response and neurogenic inflammation 51.
5. Long term undesired effects of OXA administration:
A prospective clinical study assessed OXA-treated patients with the objective of defining the prevalence, clinical impression, electrophysiological pattern and long-standing consequence of chronic OIPN by 12 months after termination of therapy. 69 patients with OXA usage for colon cancer were included in a follow-up study prospectively preceding chemotherapy and subsequent to 12 cycles of chemotherapy. Although the conditions related to OIPN improved after OXA withdrawal, more than 50% of the patients suffered from OIPN even after 12 months of withdrawing 13.
A clinical trial sought to find the potential reversibility of chronic neurotoxicity induced by OXA up to 2 years of follow-up after the discontinuation of OXA-based chemotherapy. Ninety-one patients with CRC who received an OXA-based chemotherapy regimen enrolled for a study where the subjects underwent neurological evaluation, clinical TNSc, and nerve conduction studies at baseline (T0), after chemotherapy (T1), and 2 years after chemotherapy (T2) respectively. Significantly, 80% (73/91) patients had OIPN at T1. After a median follow-up of 25 months, 84% (61/73) of patients had sustained chronic OIPN and 12 patients (17%) had complete remission. A longitudinal comparison of TNSc values between T1 and T2 found that the overall severity of OIPN in 61 patients decreased significantly over time. The mean TNSc values were nine (range: 2-15, T1) and four (range: 2-12, T2) (p < 0.001). It is common to find that OIPN lasts for more than 2 years after chemotherapy is completed 52.
Therefore, OXA results in chronic neurotoxicity, which may affect the patients over a longer course of exposure resulting in a serious decline in the quality of their life.
6. Treatment options for OIPN:
At present, chronic OIPN has no recognized preventive measures. But the general treatment is mostly aimed to reduce the dose of OXA by 25% once the sensation abnormality persists in the intermittent period. If dysfunction occurs, the drug will be discontinued, which will eventually affect its anti-tumor effect 53. Significant agents for management of OIPN and their mechanisms of action have been presented in this section.
6.1. Duloxetine:
6.1.1. Preclinical trials:
Duloxetine is a serotonin and a noradrenaline re-uptake inhibitor, which remains a first choice medication recommended by American Society of Clinical Oncology (ASCO) guidelines for the treatment of chemotherapy-induced peripheral neuropathic pain. In terms of the mechanisms of action engaged, it is widely acclaimed that duloxetine reduces the inflammation and nerve injury by suppressing p38 phosphorylation activation and its downstream NF-κB nuclear translocation 54. Kim, et al., reported that duloxetine (30 mg/kg) could remarkably suppress allodynia and the excessive excitability of spinal wide dynamic range (WDR) neurons facilitated by spinal α₁-adrenergic receptors. This mode of action of duloxetine effectively reduced the level of pain exacerbated by the administration of the chemotherapeutic agent 55. In a mice model for neuropathy, duloxetine improved the OXA-induced changes significantly by impeding the p53-linked apoptosis. In short, the drug was effective in alleviating the alterations in thermal withdrawal latency, paw withdrawal threshold and intraepidermal nerve fiber density 56. Furthermore, duloxetine alleviated the symptoms of allodynia by preventing the phosphorylation of ERK1/2 in the spinal cords of neuropathy-induced mice 57.
6.1.2. Clinical trials:
ASCO has suggested that duloxetine may be beneficial for the treatment and prevention of OIPN, based on several randomized clinical trials 58. For instance, a randomly Phase III double-blind clinical trial enrolled 231 patients to assess whether duloxetine at the dose of 60 mg every day can decrease the symptoms of grade 1 OIPN. The treatment with duloxetine indicated a larger decrease in average pain induced by OXA with improved function, quality of life (QOL) and limited side effects 59. Although strong therapeutic assumptions were not derived out of a randomized, placebo-controlled, double-blind clinical trial after 14 days of administering duloxetine, the drug was considerably safe and presented better physiological tolerance in rectal cancer patients 60. In a yet another open-label pilot study, thirty-four cancer patients randomly received duloxetine (40 mg/day) or vitamin B12 (1.5 mg/day) orally for 4 weeks. The pain and numbness scores were significantly decreased after treatment with duloxetine, compared to vitamin B12 treatment. Fatigue was the most common side effect observed 61. Another trial enrolled thirty-nine colorectal cancer patients with chronic OIPN. After 12 weeks of administering duloxetine (60 mg/day), the pain was evaluated by visual analog scale (VAS) score. The outcomes indicated that over 63% patients got a VAS score improvement without significant renal or hepatic damage. The dose presented limited or acceptable levels of side effects 62.
Remarkably, these studies not only provided abundant evidence to support the use of duloxetine as a standard OIPN drug, but also indicated the potential targets for OIPN drug development.
6.2. Niclosamide:
6.2.1. Preclinical studies:
There is a comprehensive study that reported potential anti-OIPN and neuroprotective effects of niclosamide at preclinical level against OXA-induced OIPN. In vitro, normal neuron-like and cancer cells as well as mice grafted with CT26 colon cancer cells in vivo were used. The cytotoxic effects of OXA were promoted by niclosamide specifically on CT26 and THP1 cells, whereas, the toxic effects on normal neuron-like cells were diminished. Niclosamide downregulated the production of H2O2 mediated by OXA among the normal cells used, thereby preventing cell death among such cells. However, in colon cancer cells, niclosamide enhanced OXA-mediated cell death through increased H2O2 production. In a mice model for neuropathy, niclosamide assisted in avoiding tactile hypoesthesia and thermal hyperalgesia besides resulting in an revoked membrane hyperexcitability. This report pointed out that niclosamide employs its neuroprotective properties both atcellular levelsand at tissue levels in animals by controlling oxidative stress and neuroinflammation induced by OXA. The conclusions of this study distinguish niclosamide as a capable therapeutic adjunct to chemotherapeutic approach using OXA 63.
6.2.2. Clinical trials:
Till date, there are no clinical reports on the management of OIPN using niclosamide. The preclinical report mentioned above can provide supportive evidence for the use of niclosamide at clinical levels after further collective research.
6.3. Glutathione:
6.3.1. Preclinical studies:
Cystine/Theanine as the precursors of the biosynthesis of glutathione, can significantly increase the glutathione levels, which is beneficial for mechanical allodynia and axonal degeneration induced by OXA 64. Studies also found that increasing the activity of glutathione in brain tissue by using drug possibilities for cancers may help in enhancing the beneficial effects on OIPN 65.
6.3.2. Clinical trials:
A randomized, double-blind, placebo-controlled trial was performed to assess the efficacy of glutathione (GSH) in the prevention of OIPN. A total of 52 patients treated with a OXA-based regimen for every two weeks, selected and separated to receive GSH (1,500 mg/m2 across a 15-minute exposure before OXA). After 12 cycles, grade 2 to 4 neurotoxicity was observed among three patients in the GSH arm and eight patients in the placebo arm. The neurophysiologic examination displayed a substantial decrease of the values in the placebo arm but not in the GSH arm. GSH can improve the therapeutic index of OXA and help in reducing OIPN 66. Other evidence also proved the beneficial of some amino acids which are related to glutathione in clinic. In an attempt of illustrating such benefits, Kobayashi et al., used a grading scale for adverse events to assess the efficacy and toxicity profile of glutathione. The results showed that oral administration of GSH precursors every day attenuated OIPN, which is consistent with basic studies 67. Similarly, N-acetylcysteine as a precursor for glutathione increases the blood levels of the antioxidant. It has been found that orally administering colorectal or gastric cancer patients with N-acetylcysteine (1200 mg) 1 hour before receiving OXA can reduce the incidence of OIPN based on the results of neuropathy grading and electrophysiological assessments 68. This study suggests that glutathione is a promising agent for the prevention and in delaying the onset of OIPN.
6.4. alpha-lipoic acid:
6.4.1. Preclinical studies:
Chemotherapeutic drugs cause peripheral neuropathy by increasing ROS, leading to the activation of ion channel TRPA1 via nociceptors. Alpha-lipoic acid (ALA) as an oxidative stress scavenger, the effect in OIPN has been explored recently. In animal experiments, it has been reported that ALA could completely prevent ROS that could be generated as a product of hypersensitivity. If administered prior to therapeutic drug it can inhibit TRPA1-induced hypersensitivity and prevent OIPN 69. Another in vivo study showed that the ALA had protective effects on OXA-induced hyperalgesia in rats 70. In tumor-bearing mice, OXA increased the production of ROS, augmented the content of pro-inflammatory cytokines (IL-1β and TNF-α) and astrocytic markers of the spinal cord. Interestingly, ALA inhibited the ROS-dependent neuroinflammation, leading to the alleviation of OIPN 71.
6.4.2. Clinical trials:
ALA was found to be effective in a small open-label pilot study when co-administered with OXA. The treatment combination used in this pilot study (600 mg ALA given intravenously once a week for 3–5 weeks followed by 600 mg three times daily orally) showed a trend toward a reduction in the severity of OIPN among 8 out of the 15 patients 72. Another clinical study included 70 patients who received 600 mg of ALA or placebo three times a day for 24 weeks and completed the trial. Yet, the results showed that the neurotoxicity grades of the two groups were both decreased. Significant differences among both the groups in the preventing or treating OIPN, or a reduction in Brief Pain Inventory (BPI) score and functional decline were not observed. Moreover, it was to be noted that in this trial, the treatment validated a poor patient compliance resulting in high wastage rate and poor feasibility 73. Therefore, ALA is a translational potential compound for management of OIPN at the clinical level.
6.5. Mangafodipir:
6.5.1. Preclinical studies:
The manganese chelate and superoxide dismutase mimetic mangafodipir is also an efficacious inhibitor of OIPN similar to other agents reviewed. After undergoing certain metabolic processes, mangafodipir tends to attack cellular oxidative stress at three levels to prevent OIPN without adverse events and reducing the detrimental effects of OXA 74. Coriat et al., established an animal model of C57BL/6 mice for neurotoxicity induced by OXA in a study. Electrophysiological tests indicated that mangafodipir was effective in preventing OXA-treated mice from acquiring motor and sensitivity troubles in consort with neuromuscular hyperexcitability episodes 47.
6.5.2. Clinical trials:
Coriat’s team also performed a phase II trial which enrolled 22 cancer patients treated with intravenous injection of mangafodipir. After 4 cycles, 77% of patients had alleviated symptoms of neuropathy. After 8 cycles, 6 out of 7 patients had degraded neuropthy symptoms of grade 2 47. According to these evidences and outcomes, mangafodipir is a potential and progressive agent for treating or even curing OIPN.
6.6. Calmangafodipir:
6.6.1. Preclinical studies:
Calmangafodipir as the successor of mangafodipir possesses better therapeutic activity. In an OIPN mouse model, the intraepidermal nerve fiber (IENF) density was assessed after administration of calmangafodipir followed by OXA which indicated the prevention of OIPN depending on the concentrations of calmangafodipir 75. Another study exploring the use of CT26 tumor–bearing mice also proved the therapeutic effects of calmangafodipir compared to mangafodipir. According to these results, calmangafodipir conferred significant protection against tissue damage induced by chemotherapy 76.
6.6.2. Clinical trials:
Calmangafodipir was used in a double-blinded randomised phase II study in patients with metastatic CRC at a dose of 5 µmol/kg. The drug resulted in lessened effects of cold allodynia and sensory symptoms besides causing a delay in the onset and severity of OIPN. The tumor outcomes were not influenced by the mangafodipir-mimetic 77.
6.7. Venlafaxine:
6.7.1. Preclinical studies:
Venlafaxine is a serotonin-norepinephrine reuptake inhibitor (SNRI) with analgesic effects observed in OIPN pain mice model. After administrating venlafaxine intraperitoneally, the scores of cold and mechanical allodynia were tested. An improving effect was reported after treatment with venlafaxine (40 mg/kg), suggesting the potential of venlafaxine against OIPN via the inhibition of cold and mechanical allodynia 78. Similarly, an in vivo microdialysis experiment showed that venlafaxine (16 mg/kg) overturned cold allodynia in OIPN mouse model 79.
6.7.2. Clinical trials:
Additionally, a series of case studies had supported the use of venlafaxine for prevention and treatment of acute OIPN 80-82. A small randomized placebo-controlled phase III trial (EFFOX) led by Durand et al. enrolled forty-eight patients who had reported distressing acute neurotoxicity from OXA. Significant improvements were reported for both acute and grade 3 chronic neuropathy symptoms in those who received venlafaxine, and the mechanism may be related to JNK signaling pathway 83. In yet another randomized, double-blind, placebo-controlled, pilot clinical trial using venlafaxine against OIPN reported some interesting results. 50 patients with stage 2 to 4 colon cancer who were treated with a OXA-based regimen received venlafaxine (37.5 mg) twice every day. The venlafaxine treatment group showed a better trend in the occurrence of acute neurotoxicity, but only of the initial 1-2 cycles. Other evaluations did not show this benefit. Therefore, this study does not recommend venlafaxine to prevent OIPN, nor does it recommend continuing venlafaxine for phase III clinical trials 84.
6.8. Gabapentin:
6.8.1. Preclinical studies:
Gabapentin is widely used in the treatment of neuropathic pain and neuropathic pain caused by tumors, which may benefit the peripheral nerves through the antagonism of NMDA receptors and central nervous system calcium channels 85. This mechanism of action theoretically corresponds to one of the possible mechanisms of OXA. Ohsawa et al., found that gabapentin can prevent or inhibit OXA-induced mechanical hyperalgesia, but does not influence cold allodynia in vivo 86. Another experiment showed that the cold and mechanical allodynia induced by OXA can be avoided by inhibiting ERK1/2 phosphorylation 57. Recently, gabapentin has been used as a positive control in OIPN model to evaluate the relapse of chronic sensory dysfunction caused by the administration of mesenchymal stem/stromal cells 87,88. Therefore, gabapentin could be considered as a potential anti-OIPN agent for clinical use.
Clinical trials:
In a clinical study by Zhang et al. (2015), 45 patients with colorectal cancer (CRC) who experienced grade 2-3 neurotoxicity during oxaliplatin (OXA) treatment were given gabapentin to manage their neurotoxicity. The maximum tolerated dose of gabapentin was 900 mg three times daily (tid), and sensory neurotoxicity was evaluated at various dosage levels. The findings indicated that a dose of 300 mg tid provided the most effective therapeutic benefit, although patients also showed improvement at doses below this level. Nearly half of the patients experienced a reduction in neurotoxicity to grades 1-2.
Conversely, a phase III randomized, double-blind, placebo-controlled crossover trial (N00C3) involving 115 patients with drug-induced neuropathic pain found that gabapentin did not produce significant therapeutic effects. However, the chemotherapy agents used by patients in this trial were varied and not limited to oxaliplatin.
6.9. Carbamazepine:
6.9.1. Preclinical studies:
Carbamazepine is a commonly used anticonvulsant drug, which binds to the receptor sites to block the voltage-sensitive sodium channels. In N18 cells, the inhibitory effects of carbamazepine on 22Na+ flux was shifted to elevated concentrations when measured in the existence of sodium and calcium ions associated with the function of central nervous system. This resembles the neuroprotective effects of carbamazepine 91. Exposure to OXA (250 μmol), changed the sodium channel inactivation kinetics on rat sensory sural nerve preparations. Carbamazepine can effectively prevent multiple endplate potentials (EPPs) and cause a decrease in spontaneous activity by blocking sodium channels that happened due to OXA exposure (0.5 mM) 92,93.
As a collective input, the treatment options for OIPN and the mechanisms involved are elucidated in Figure 2 and Figure 3.
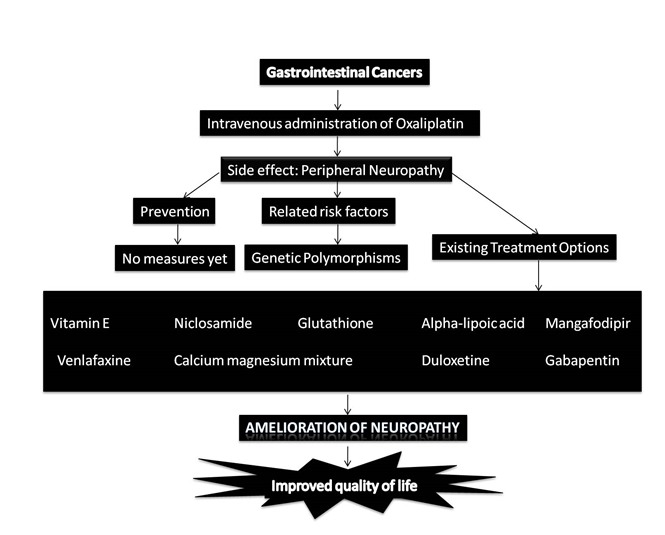
Figure 2: Factors involved in OIPN and the treatment options for improved quality of life among cancer patients.
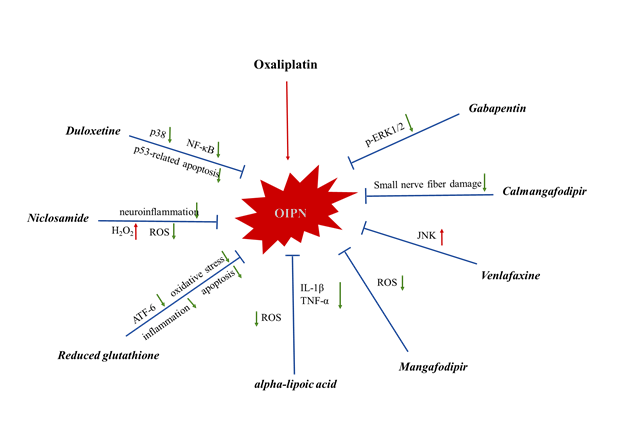
Figure 3: The mechanisms of action of anti-OIPN agents.
6.9.2. Clinical trials:
A randomised, controlled, multicenter phase II study was performed to assess the effect of carbamazepine (3 to 6 mg/l of serum) in advanced CRC patients treated with OXA. The outcomes indicated that carbamazepine can prevent OIPN of grades greater than 1 94.
7. Biased effects of agents for the management of OIPN:
7.1. Vitamin E:
Vitamin E was initially considered the first compound capable of preventing chemotherapy-related neurotoxicity. A clinical trial was conducted to assess the effect of Vitamin E on preventing oxaliplatin-induced peripheral neuropathy (OIPN) in colorectal cancer (CRC) patients. In this trial, 32 patients were assigned to the Vitamin E group, while 33 were in the control group. After the sixth cycle of the oxaliplatin-based regimen, no significant difference was observed in the mean change of peripheral neuropathy scores between the two groups (mean change in Vitamin E group = 6.37 ± 2.85; control group = 6.57 ± 2.94; P = 0.78). The study concluded that Vitamin E did not provide a benefit in preventing OIPN.
Another randomized, double-blind, placebo-controlled phase III trial enrolled 189 evaluable patients receiving chemotherapy (including paclitaxel, cisplatin, carboplatin, and oxaliplatin) along with 400 mg/day of Vitamin E; among them, 50 patients (26%) were treated with oxaliplatin. The incidence of neurotoxicity above grade 2 was similar between the Vitamin E and placebo groups (34% vs. 29%).
Overall, these studies indicate that Vitamin E does not appear to reduce the risk of chemotherapy-induced neurotoxicity.
7.2. Calcium-magnesium mixture:
A meta-analysis reported that the relative risk (OR) of the calcium-magnesium mixture compared to placebo was 0.44 (95% CI: 0.23–0.85, p=0.01), while the odds ratio (OR) for acute neurotoxicity was 0.41 (95% CI: 0.11–1.49, p=0.18). These findings suggest that the calcium-magnesium mixture may reduce the occurrence of acute peripheral neurotoxicity caused by oxaliplatin (OXA) when used as an adjunct therapy [97]. Additionally, this mixture proved effective in managing OXA-induced neuropathy at a dose of 85 mg/m² [98].
However, Wu et al. (2012) conducted a meta-analysis of seven studies, including four randomized controlled trials and three cohort studies with a total of 1,238 patients. Their results indicated that the calcium-magnesium mixture neither prevented nor decreased OXA-related neurotoxicity but did not compromise the therapeutic effectiveness of OXA-based treatments. Consequently, this meta-analysis does not support the idea that calcium-magnesium mixtures reduce the incidence of oxaliplatin-induced peripheral neuropathy (OIPN) in colorectal cancer (CRC) patients [99]. Moreover, several other studies advise against the routine use of calcium-magnesium mixtures for treating OXA-induced neurotoxicity [100,101].
7. Conclusions:
OXA, a chemotherapy drug used to treat colorectal cancer (CRC), can cause peripheral neuropathy, for which there are currently no well-established prevention or treatment options. Chronic neuropathy induced by OXA also lacks recognized preventive measures. However, several risk factors linked to OXA use mainly lead to damage in the limbs. In summary, this review highlights that although numerous drugs are clinically employed to prevent or manage OXA-induced neurotoxicity at the molecular level, none have yet received definitive recommendations. Therefore, more research is needed to determine effective treatment protocols for OXA-induced neurotoxicity.
References:
1. Lovejoy KS, Serova M, Bieche I, et al. Spectrum of cellular responses to pyriplatin, a monofunctional cationic antineoplastic platinum (II) compound, in human cancer cells. Molecular cancer therapeutics. 2011:molcanther. 0250.2011.
2. Johnstone TC. The Crystal Structure of Oxaliplatin: A Case of Overlooked Pseudo Symmetry. Polyhedron. 2014;67:10.1016/j.poly.2013.1010.1003.
3. Mehmood RK. Review of Cisplatin and oxaliplatin in current immunogenic and monoclonal antibody treatments. Oncology reviews. 2014;8(2):256-256.
4. Martinez-Balibrea E, Martínez-Cardús A, Ginés A, et al. Tumor-related molecular mechanisms of oxaliplatin resistance. Molecular cancer therapeutics. 2015;14(8):1767-1776.
5. Graham MA, Lockwood GF, Greenslade D, Brienza S, Bayssas M, Gamelin E. Clinical pharmacokinetics of oxaliplatin: a critical review. Clinical cancer research : an official journal of the American Association for Cancer Research. Apr 2000;6(4):1205-1218.
6. Cersosimo RJ. Oxaliplatin-associated neuropathy: a review. Annals of Pharmacotherapy. 2005;39(1):128-135.
7. Sałat K. Chemotherapy-induced peripheral neuropathy—part 2: focus on the prevention of oxaliplatin-induced neurotoxicity. Pharmacological Reports. 2020;72(3):508-527.
8. Grolleau Fo, Gamelin L, Boisdron-Celle Ml, Lapied B, Pelhate M, Gamelin E. A possible explanation for a neurotoxic effect of the anticancer agent oxaliplatin on neuronal voltage-gated sodium channels. Journal of neurophysiology. 2001;85(5):2293-2297.
9. Kang L, Tian Y, Xu S, Chen H. Oxaliplatin-induced peripheral neuropathy: clinical features, mechanisms, prevention and treatment. Journal of Neurology. 2021;268(9):3269-3282.
10. Piccolo J, Kolesar JM. Prevention and treatment of chemotherapy-induced peripheral neuropathy. American journal of health-system pharmacy. 2014;71(1):19-25.
11. Yang Y, Zhao B, Gao X, et al. Targeting strategies for oxaliplatin-induced peripheral neuropathy: clinical syndrome, molecular basis, and drug development. Journal of Experimental & Clinical Cancer Research. 2021;40(1):1-25.
12. Ewertz M, Qvortrup C, Eckhoff L. Chemotherapy-induced peripheral neuropathy in patients treated with taxanes and platinum derivatives. Acta oncologica. 2015;54(5):587-591.
13. Kim SH, Kim W, Kim JH, et al. A Prospective Study of Chronic Oxaliplatin-Induced Neuropathy in Patients with Colon Cancer: Long-Term Outcomes and Predictors of Severe Oxaliplatin-Induced Neuropathy. J Clin Neurol. 2018;14(1):81-89.
14. Grothey A. Clinical management of oxaliplatin-associated neurotoxicity. Clinical colorectal cancer. 2005;5:S38-S46.
15. Hoff PM, Saad ED, Costa F, et al. Literature review and practical aspects on the management of oxaliplatin-associated toxicity. Clinical colorectal cancer. 2012;11(2):93-100.
16. Argyriou AA, Kyritsis AP, Makatsoris T, Kalofonos HP. Chemotherapy-induced peripheral neuropathy in adults: a comprehensive update of the literature. Cancer management and research. 2014;6:135-147.
17. Beijers A, Mols F, Vreugdenhil G. A systematic review on chronic oxaliplatin-induced peripheral neuropathy and the relation with oxaliplatin administration. Supportive Care in Cancer. 2014;22(7):1999-2007.
18. Ducreux M, Bennouna J, Hebbar M, et al. Capecitabine plus oxaliplatin (XELOX) versus 5‐fluorouracil/leucovorin plus oxaliplatin (FOLFOX‐6) as first‐line treatment for metastatic colorectal cancer. International journal of cancer. 2011;128(3):682-690.
19. Argyriou A, Velasco R, Briani C, et al. Peripheral neurotoxicity of oxaliplatin in combination with 5-fluorouracil (FOLFOX) or capecitabine (XELOX): a prospective evaluation of 150 colorectal cancer patients. Annals of oncology. 2012;23(12):3116-3122.
20. Besora S, Santos C, Izquierdo C, Martinez-Villacampa MM, Bruna J, Velasco R. Rechallenge with oxaliplatin and peripheral neuropathy in colorectal cancer patients. Journal of cancer research and clinical oncology. 2018;144(9):1793-1801.
21. Argyriou AA, Antonacopoulou AG, Scopa CD, et al. Liability of the voltage-gated sodium channel gene SCN2A R19K polymorphism to oxaliplatin-induced peripheral neuropathy. Oncology. 2009;77(3-4):254-256.
22. Argyriou AA, Cavaletti G, Antonacopoulou A, et al. Voltage‐gated sodium channel polymorphisms play a pivotal role in the development of oxaliplatin‐induced peripheral neurotoxicity: Results from a prospective multicenter study. Cancer. 2013;119(19):3570-3577.
23. Palugulla S, Thakkar DN, Kayal S, Narayan SK, Dkhar SA. Association of Voltage-Gated Sodium Channel Genetic Polymorphisms with Oxaliplatin-Induced Chronic Peripheral Neuropathy in South Indian Cancer Patients. Asian Pacific journal of cancer prevention: APJCP. 2017;18(11):3157.
24. Kitade H, Shimasaki T, Igarashi S, et al. Long‑term administration and efficacy of oxaliplatin with no neurotoxicity in a patient with rectal cancer: Association between neurotoxicity and the GSTP1 polymorphism. Oncology letters. 2014;7(5):1499-1502.
25. Peng Z, Wang Q, Gao J, et al. Association between GSTP1 Ile105Val polymorphism and oxaliplatin-induced neuropathy: a systematic review and meta-analysis. Cancer chemotherapy and pharmacology. 2013;72(2):305-314.
26. Won HH, Lee J, Park JO, et al. Polymorphic markers associated with severe oxaliplatin‐induced, chronic peripheral neuropathy in colon cancer patients. Cancer. 2012;118(11):2828-2836.
27. Terrazzino S, Argyriou AA, Cargnin S, et al. Genetic determinants of chronic oxaliplatin‐induced peripheral neurotoxicity: a genome‐wide study replication and meta‐analysis. Journal of the Peripheral Nervous System. 2015;20(1):15-23.
28. Kanai M, Kawaguchi T, Kotaka M, et al. Large-scale prospective pharmacogenomics study of oxaliplatin-induced neuropathy in colon cancer patients enrolled in the JFMC41-1001-C2 (JOIN Trial). Annals of Oncology. 2016;27(6):1143-1148.
29. Gamelin L, Capitain O, Morel A, et al. Predictive factors of oxaliplatin neurotoxicity: the involvement of the oxalate outcome pathway. Clinical Cancer Research. 2007;13(21):6359-6368.
30. Custodio A, Moreno-Rubio J, Aparicio J, et al. Pharmacogenetic predictors of severe peripheral neuropathy in colon cancer patients treated with oxaliplatin-based adjuvant chemotherapy: a GEMCAD group study. Annals of oncology. 2013;25(2):398-403.
31. Oguri T, Mitsuma A, Inada-Inoue M, et al. Genetic polymorphisms associated with oxaliplatin-induced peripheral neurotoxicity in Japanese patients with colorectal cancer. International journal of clinical pharmacology and therapeutics. 2013;51(6):475-481.
32. Cliff J, Jorgensen A, Lord R, et al. The molecular genetics of chemotherapy–induced peripheral neuropathy: A systematic review and meta-analysis. Critical reviews in oncology/hematology. 2017.
33. Liu C-N, Michaelis M, Amir R, Devor M. Spinal nerve injury enhances subthreshold membrane potential oscillations in DRG neurons: relation to neuropathic pain. Journal of Neurophysiology. 2000;84(1):205-215.
34. Anand U, Otto WR, Anand P. Sensitization of capsaicin and icilin responses in oxaliplatin treated adult rat DRG neurons. Molecular pain. 2010;6(1):82.
35. Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA base excision repair protein, APE1 in cisplatin, oxaliplatin, or carboplatin induced sensory neuropathy. PloS one. 2014;9(9):e106485.
36. McKeage M, Hsu T, Screnci D, Haddad G, Baguley B. Nucleolar damage correlates with neurotoxicity induced by different platinum drugs. British journal of cancer. 2001;85(8):1219.
37. Jamieson SM, Subramaniam J, Liu JJ, et al. Oxaliplatin-induced loss of phosphorylated heavy neurofilament subunit neuronal immunoreactivity in rat DRG tissue. Molecular Pain. 2009;5(1):66.
38. Cavaletti G, Miloso M, Nicolini G, Scuteri A, Tredici G. Emerging role of mitogen‐activated protein kinases in peripheral neuropathies. Journal of the Peripheral Nervous System. 2007;12(3):175-194.
39. Ma Q-P, Hargreaves R. Localization of N-methyl-D-aspartate NR2B subunits on primary sensory neurons that give rise to small-caliber sciatic nerve fibers in rats. Neuroscience. 2000;101(3):699-707.
40. Mihara Y, Egashira N, Sada H, et al. Involvement of spinal NR2B-containing NMDA receptors in oxaliplatin-induced mechanical allodynia in rats. Molecular pain. 2011;7(1):8.
41. Chen Y, Yang C, Wang Z. Proteinase-activated receptor 2 sensitizes transient receptor potential vanilloid 1, transient receptor potential vanilloid 4, and transient receptor potential ankyrin 1 in paclitaxel-induced neuropathic pain. Neuroscience. 2011;193:440-451.
42. Ta LE, Bieber AJ, Carlton SM, Loprinzi CL, Low PA, Windebank AJ. Transient Receptor Potential Vanilloid 1 is essential for cisplatin-induced heat hyperalgesia in mice. Molecular pain. 2010;6:1744-8069-1746-1715.
43. Gauchan P, Andoh T, Kato A, Kuraishi Y. Involvement of increased expression of transient receptor potential melastatin 8 in oxaliplatin-induced cold allodynia in mice. Neuroscience letters. 2009;458(2):93-95.
44. Krishnan AV, Goldstein D, Friedlander M, Kiernan MC. Oxaliplatin‐induced neurotoxicity and the development of neuropathy. Muscle & Nerve: Official Journal of the American Association of Electrodiagnostic Medicine. 2005;32(1):51-60.
45. Donzelli E, Carfì M, Miloso M, et al. Neurotoxicity of platinum compounds: comparison of the effects of cisplatin and oxaliplatin on the human neuroblastoma cell line SH-SY5Y. Journal of Neuro-oncology. 2004;67(1-2):65-73.
46. Waseem M, Kaushik P, Tabassum H, Parvez S. Role of mitochondrial mechanism in chemotherapy-induced peripheral neuropathy. Current drug metabolism. 2018;19(1):47-54.
47. Coriat R, Alexandre J, Nicco C, et al. Treatment of oxaliplatin-induced peripheral neuropathy by intravenous mangafodipir. The Journal of clinical investigation. 2014;124(1):262-272.
48. Carozzi V, Marmiroli P, Cavaletti G. The role of oxidative stress and anti-oxidant treatment in platinum-induced peripheral neurotoxicity. Current cancer drug targets. 2010;10(7):670-682.
49. Carozzi V, Canta A, Chiorazzi A. Chemotherapy-induced peripheral neuropathy: what do we know about mechanisms? Neuroscience letters. 2015;596:90-107.
50. Nassini R, Gees M, Harrison S, et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. PAIN®. 2011;152(7):1621-1631.
51. Lim SC, Choi JE, Kang HS, Si H. Ursodeoxycholic acid switches oxaliplatin‐induced necrosis to apoptosis by inhibiting reactive oxygen species production and activating p53‐caspase 8 pathway in HepG2 hepatocellular carcinoma. International journal of cancer. 2010;126(7):1582-1595.
52. Briani C, Argyriou AA, Izquierdo C, et al. Long‐term course of oxaliplatin‐induced polyneuropathy: a prospective 2‐year follow‐up study. Journal of the Peripheral Nervous System. 2014;19(4):299-306.
53. Cao P, Yang J, Cai X, Wang X, Huo J. Treatment of chemotherapy-related peripheral neuropathy with traditional Chinese medicine from the perspective of blood-arthralgia Zheng. TANG [HUMANITAS MEDICINE]. 2012;2(4):30.31-30.34.
54. Meng J, Zhang Q, Yang C, Xiao L, Xue Z, Zhu J. Duloxetine, a balanced serotonin-norepinephrine reuptake inhibitor, improves painful chemotherapy-induced peripheral neuropathy by inhibiting activation of p38 MAPK and NF-κB. Frontiers in pharmacology. 2019;10:365.
55. Kim W, Chung Y, Choi S, Min B-I, Kim SK. Duloxetine protects against oxaliplatin-induced neuropathic pain and spinal neuron hyperexcitability in rodents. Int J Mol Sci. 2017;18(12):2626.
56. Wang M, Zhang L, Liu X, et al. Duloxetine alleviates oxaliplatin-induced peripheral neuropathy by regulating p53-mediated apoptosis. NeuroReport. 2022;33(10):437-444.
57. Kato N, Tateishi K, Tsubaki M, et al. Gabapentin and Duloxetine Prevent Oxaliplatin-and Paclitaxel-Induced Peripheral Neuropathy by Inhibiting Extracellular Signal-Regulated Kinase 1/2 (ERK1/2) Phosphorylation in Spinal Cords of Mice. Pharmaceuticals. 2020;14(1):30.
58. Hershman DL, Lacchetti C, Loprinzi CL. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline summary. Journal of oncology practice. 2014;10(6):e421-e424.
59. Smith EML, Pang H, Cirrincione C, et al. Effect of duloxetine on pain, function, and quality of life among patients with chemotherapy-induced painful peripheral neuropathy: a randomized clinical trial. Jama. 2013;309(13):1359-1367.
60. Aghili M, Darzikolaee NM, Babaei M, et al. Duloxetine for the prevention of oxaliplatin induced peripheral neuropathy: a randomized, placebo-controlled, double-blind clinical trial. Journal of Gastrointestinal Cancer. 2022:1-8.
61. Hirayama Y, Ishitani K, Sato Y, et al. Effect of duloxetine in Japanese patients with chemotherapy-induced peripheral neuropathy: a pilot randomized trial. International journal of clinical oncology. 2015;20(5):866-871.
62. Yang Y-H, Lin J-K, Chen W-S, et al. Duloxetine improves oxaliplatin-induced neuropathy in patients with colorectal cancer: an open-label pilot study. Supportive Care in Cancer. 2012;20(7):1491-1497.
63. Cerles O, Benoit E, Chéreau C, et al. Niclosamide inhibits oxaliplatin neurotoxicity while improving colorectal cancer therapeutic response. Molecular cancer therapeutics. 2016:molcanther. 0326.2016.
64. Kawashiri T, Kobayashi D, Egashira N, Tsuchiya T, Shimazoe T. Oral administration of Cystine and Theanine ameliorates oxaliplatin-induced chronic peripheral neuropathy in rodents. Scientific reports. 2020;10(1):1-8.
65. Celik H, Kucukler S, Ozdemir S, et al. Lycopene protects against central and peripheral neuropathy by inhibiting oxaliplatin-induced ATF-6 pathway, apoptosis, inflammation and oxidative stress in brains and sciatic tissues of rats. Neurotoxicology. 2020;80:29-40.
66. Cascinu S, Catalano V, Cordella L, et al. Neuroprotective effect of reduced glutathione on oxaliplatin-based chemotherapy in advanced colorectal cancer: a randomized, double-blind, placebo-controlled trial. Journal of Clinical Oncology. 2002;20(16):3478-3483.
67. Kobayashi M, Sato R, Komura T, et al. Protective effect of the oral administration of cystine and theanine on oxaliplatin-induced peripheral neuropathy: A pilot randomized trial. International journal of clinical oncology. 2020;25(10):1814-1821.
68. Bondad N, Boostani R, Barri A, Elyasi S, Allahyari A. Protective effect of N-acetylcysteine on oxaliplatin-induced neurotoxicity in patients with colorectal and gastric cancers: a randomized, double blind, placebo-controlled, clinical trial. Journal of Oncology Pharmacy Practice. 2020;26(7):1575-1582.
69. Trevisan G, Materazzi S, Fusi C, et al. Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Research. 2013;73(10):3120-3131.
70. Joseph EK, Chen X, Bogen O, Levine JD. Oxaliplatin acts on IB4-positive nociceptors to induce an oxidative stress-dependent acute painful peripheral neuropathy. The journal of pain. 2008;9(5):463-472.
71. Agnes JP, Dos Santos VW, das Neves RN, et al. Antioxidants improve oxaliplatin-induced peripheral neuropathy in tumor-bearing mice model: role of spinal cord oxidative stress and inflammation. The Journal of Pain. 2021;22(8):996-1013.
72. Gedlicka C, Scheithauer W, Schull B, Kornek G. Effective treatment of oxaliplatin-induced cumulative polyneuropathy with alpha-lipoic acid. Journal of clinical oncology. 2002;20(15):3359-3361.
73. Guo Y, Jones D, Palmer JL, et al. Oral alpha-lipoic acid to prevent chemotherapy-induced peripheral neuropathy: a randomized, double-blind, placebo-controlled trial. Supportive Care in Cancer. 2014;22(5):1223-1231.
74. Karlsson JOG, Andersson RG, Jynge P. Mangafodipir a selective cytoprotectant—with special reference to oxaliplatin and its association to chemotherapy-induced peripheral neuropathy (CIPN). Translational Oncology. 2017;10(4):641-649.
75. Canta A, Chiorazzi A, Pozzi E, et al. Calmangafodipir reduces sensory alterations and prevents intraepidermal nerve fibers loss in a mouse model of oxaliplatin induced peripheral neurotoxicity. Antioxidants. 2020;9(7):594.
76. Karlsson JOG, Kurz T, Flechsig S, Näsström J, Andersson RG. Superior therapeutic index of calmangafodipir in comparison to mangafodipir as a chemotherapy adjunct. Translational oncology. 2012;5(6):492-502.
77. Glimelius B, Manojlovic N, Pfeiffer P, et al. Persistent prevention of oxaliplatin-induced peripheral neuropathy using calmangafodipir (PledOx((R))): a placebo-controlled randomised phase II study (PLIANT). Acta oncologica (Stockholm, Sweden). Mar 2018;57(3):393-402.
78. Li D, Lee JH, Choi CW, Kim J, Kim SK, Kim W. The analgesic effect of venlafaxine and its mechanism on oxaliplatin-induced neuropathic pain in mice. Int J Mol Sci. 2019;20(7):1652.
79. Hache G, Guiard B, Nguyen T, et al. Antinociceptive activity of the new triple reuptake inhibitor NS 18283 in a mouse model of chemotherapy‐induced neuropathic pain. European Journal of Pain. 2015;19(3):322-333.
80. Durand JP, Alexandre J, Guillevin L, Goldwasser F. Clinical activity of venlafaxine and topiramate against oxaliplatin-induced disabling permanent neuropathy. Anti-cancer drugs. Jun 2005;16(5):587-591.
81. Durand JP, Brezault C, Goldwasser F. Protection against oxaliplatin acute neurosensory toxicity by venlafaxine. Anti-cancer drugs. Jul 2003;14(6):423-425.
82. Özdogan M, Samur M, Bozcuk HŠ, Aydin H, Çoban E, B S. Venlafaxine for treatment of chemotherapy-induced neuropathic pain. Turkish Journal of Cancer. 2004;34(3).
83. Durand JP, Deplanque G, Montheil V, et al. Efficacy of venlafaxine for the prevention and relief of oxaliplatin-induced acute neurotoxicity: results of EFFOX, a randomized, double-blind, placebo-controlled phase III trial. Annals of oncology : official journal of the European Society for Medical Oncology. Jan 2012;23(1):200-205.
84. Zimmerman C, Atherton PJ, Pachman D, et al. MC11C4: a pilot randomized, placebo-controlled, double-blind study of venlafaxine to prevent oxaliplatin-induced neuropathy. Supportive care in cancer : official journal of the Multinational Association of Supportive Care in Cancer. Mar 2016;24(3):1071-1078.
85. Kukkar A, Bali A, Singh N, Jaggi AS. Implications and mechanism of action of gabapentin in neuropathic pain. Archives of pharmacal research. Mar 2013;36(3):237-251.
86. Ohsawa M, Otake S, Murakami T, Yamamoto S, Makino T, Ono H. Gabapentin prevents oxaliplatin-induced mechanical hyperalgesia in mice. Journal of pharmacological sciences. 2014;125(3):292-299.
87. Oliveira ALL, Santos GG, Espirito-Santo RF, et al. Reestablishment of redox homeostasis in the nociceptive primary afferent as a mechanism of antinociception promoted by mesenchymal stem/stromal cells in oxaliplatin-induced chronic peripheral neuropathy. Stem Cells International. 2021;2021.
88. Dos Santos GGL, Oliveira ALL, Santos DS, et al. Mesenchymal stem cells reduce the oxaliplatin-induced sensory neuropathy through the reestablishment of redox homeostasis in the spinal cord. Life Sciences. 2021;265:118755.
89. Zhang F, Yi FT, Xie JJ, Shao ZX, Xu ZH, Zhang G. Effect of gabapentin on neurotoxicity of oxaliplatin in colorectal cancer patients. Chinese Journal of New Drugs. 04/30 2015;24:904-907.
90. Rao RD, Michalak JC, Sloan JA, et al. Efficacy of gabapentin in the management of chemotherapy-induced peripheral neuropathy: a phase 3 randomized, double-blind, placebo-controlled, crossover trial (N00C3). Cancer. Nov 1 2007;110(9):2110-2118.
91. Willow M, Kuenzel E, Catterall W. Inhibition of voltage-sensitive sodium channels in neuroblastoma cells and synaptosomes by the anticonvulsant drugs diphenylhydantoin and carbamazepine. Molecular pharmacology. 1984;25(2):228-234.
92. Lersch C, Schmelz R, Eckel F, et al. Prevention of oxaliplatin-induced peripheral sensory neuropathy by carbamazepine in patients with advanced colorectal cancer. Clinical Colorectal Cancer. 2002;2(1):54-58.
93. Webster RG, Brain KL, Wilson RH, Grem JL, Vincent A. Oxaliplatin induces hyperexcitability at motor and autonomic neuromuscular junctions through effects on voltage‐gated sodium channels. British journal of pharmacology. 2005;146(7):1027-1039.
94. Eckel F, Schmelz R, Adelsberger H, Erdmann J, Quasthoff S, Lersch C. Prevention of oxaliplatin-induced neuropathy by carbamazepine. A pilot study. Deutsche Medizinische Wochenschrift (1946). 2002;127(3):78-82.
95. Salehi Z, Roayaei M. Effect of Vitamin E on oxaliplatin-induced peripheral neuropathy prevention: A randomized controlled trial. International journal of preventive medicine. 2015;6.
96. Kottschade LA, Sloan JA, Mazurczak MA, et al. The use of vitamin E for the prevention of chemotherapy-induced peripheral neuropathy: results of a randomized phase III clinical trial. Supportive Care in Cancer. 2011;19(11):1769-1777.
97. Ao R, Wang Y-H, Li R-W, Wang Z-R. Effects of calcium and magnesium on acute and chronic neurotoxicity caused by oxaliplatin: A meta-analysis. Exp Ther Med. 2012;4(5):933-937.
98. Gamelin L, Boisdron-Celle M, Delva R, et al. Prevention of oxaliplatin-related neurotoxicity by calcium and magnesium infusions: a retrospective study of 161 patients receiving oxaliplatin combined with 5-Fluorouracil and leucovorin for advanced colorectal cancer. Clinical Cancer Research. 2004;10(12):4055-4061.
99. Wu Z, Ouyang J, He Z, Zhang S. Infusion of calcium and magnesium for oxaliplatin-induced sensory neurotoxicity in colorectal cancer: A systematic review and meta-analysis. European Journal of Cancer. 2012/08/01/ 2012;48(12):1791-1798.
100. Loprinzi CL, Qin R, Dakhil SR, et al. Phase III randomized, placebo-controlled, double-blind study of intravenous calcium and magnesium to prevent oxaliplatin-induced sensory neurotoxicity (N08CB/Alliance). Journal of clinical oncology : official journal of the American Society of Clinical Oncology. Apr 1 2014;32(10):997-1005.
101. Chen AS, Solimando Jr DA, Waddell JA. Gefitinib, Fluorouracil, Oxaliplatin, and Leucovorin (IFOX) Regimen for Colorectal Cancer. Hospital Pharmacy. 2013;48(11):905-911.
102. Hershman DL, Lacchetti C, Dworkin RH, et al. Prevention and management of chemotherapy-induced peripheral neuropathy in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. Jun 20 2014;32(18):1941-1967.