
Introduction
The kinetics of inhibition for nonbranched-chain processes of saturated free-radical addition to the C=C and C=O double bonds of alkene and formaldehyde molecules, respectively, by low-reactive free radicals that can experience delocalization of the unpaired p-electron was first considered in [1]. In these processes a low- reactive free radical is formed in the reaction competing with chain propagation reactions through a reactive free radical. In the present work the kinetics of inhibition by low-reactive hydrotetraoxyl free radical is considered for the nonbranched-chain process of the addition of a hydrogen atom to one of the two multiply bonded atoms of the oxygen molecule yielding a hydroperoxyl free radical. The hydroperoxyl free radical then abstracts the most labile atom from a molecule of the compound being oxidized or decomposes to turn into a molecule of an oxidation product. The only reaction that can compete with these two reactions at the chain evolution stage is the addition of the peroxyl radical to the oxygen molecule (provided that the oxygen concentration is sufficiently high). This reaction yields the secondary hydrotetraoxyl 1:2 adduct radical, which is the heaviest and the largest among the reactants. It is less reactive than the primary peroxyl 1:1 adduct radical and, as a consequence, does not participate in further chain propagation. At moderate temperatures, the reaction proceeds via a nonbranched- chain mechanism.
The aim of this study was the mathematical simulation of oxidation process autoinhibited by oxygen, when the dependence of the peroxide formation rate on the dissolved oxygen concentration has a maximum. The simulation was based on experimental data obtained for γ-radiation-induced addition reaction of hydrogen atom to the molecular oxygen for which the initiation rate V1 is known (taking into account that V = GP and V1 = G(H•)P, where P is the dose rate, and G(H•) is the initial yield of the chain-carrier hydrogen atom H• – initiation yield [2, 3]).
Based on the reaction scheme suggested for the kinetic description of the addition process to oxygen, the kinetic equations with one to three parameters to be determined directly were derived. Reducing the number of unknown parameters in a kinetic equation will allow one to decrease the narrowness of the correlation of these parameters and to avoid a sharp buildup of the statistical error in the nonlinear estimation of these parameters in the case of a limited number of experimental data points. The rate constant of the addition to oxygen, estimated as a kinetic parameter, can be compared to its reference value if the latter is known. This provides a clear criterion to validate the mathematical description against experimental data.
KiNetiCs of Hydrogen Atom Addi Tion to the Oxygen MOlecule
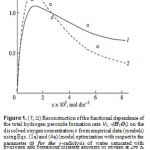
A number of experimental findings concerning the autoinhibiting effect of an increasing oxygen concentration at modest temperatures on hydrogen oxidation both in the gas phase [4–6] (Fig. 2) and in the liquid phase [7] (Fig. 1, curve 2), considered in my earlier works [8–11], can also be explained in terms of the competition kinetics of free radical addition [12, 13]. From Fig. 2 shows that the quantum yields of hydrogen peroxide and water (of products of photochemical oxidation of hydrogen at atmospheric pressure and room temperature) are maximum in the region of small concentrations of oxygen in the hydrogen–oxygen system (curves 1 and 2, respectively) [4].
In the familiar monograph “Chain Reactions” by Semenov [14], it is noted that raising the oxygen concentration when it is already sufficient usually slows down the oxidation process by shortening the chains. The existence of the upper (second) ignition limit in oxidation is due to chain termination in the bulk through triple collisions between an active species of the chain reaction and two oxygen molecules (at sufficiently high oxygen partial pressures). In the gas phase at atmospheric pressure, the number of triple collisions is roughly estimated to be 103 times smaller than the number of binary collisions (and the probability of a reaction taking place depends on the specificity of the action of the third particle) [14]. Note that in the case of a gas-phase oxidation of hydrogen at low pressures of 25-77 Pа and a temperature of 77 К [5] when triple collisions are unlikely, the dependence of the rate of hydrogen peroxide formation on oxygen concentration (the rate of passing of molecular oxygen via the reaction tube) also has a pronounced maximum (see curves 3 and 4 in Fig. 2) that indicates a chemical mechanism providing the appearance of a maximum (see reaction 4 of Scheme).
 |
 |
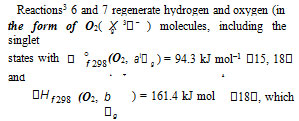
1According to Francisco and Williams [15], the enthalpy of formation (ΔНf˚298 ) in the gas phase of Н•, НО•, HO2, HO4 (the latter without the possible intramolecular hydrogen bond taken into account), О3, Н2О 6, Н2О2, and Н2О4 is 218.0 0.0, 39.0 1.2, 12.6 1.7, 122.6 13.7, 143.1 1.7, –241.8 0.0, –136.0 0, and –26.0 9 kJ mol–1, respectively. Calculations for the HO4 radical with a helical structure were carried out using the G2(MP2) method [16]. The stabilization nergies of HO2, HO4 , and HO3 were calculated in the same work to be 64.5 0.1, 69.5 0.8, and 88.5 0.8 kJ mol–1, respectively. The types of the O4 molecular dimers, their IR spectra, and higher oxygen oligomers were reported [ 17, 18]. The structure and IR spectrum of the hypothetical cyclotetraoxygen molecule O4, a species with a high energy density, were calculated by the CCSD method, and its enthalpy of formation was estimated [19]. The photochemical properties of O4 and the van der Waals nature of the О2–О2 bond were investigated [20, 21]. The most stable geometry of the dimer is two O2 molecules parallel to one another. The O4 molecule was identified by NR mass spectrometry [22].
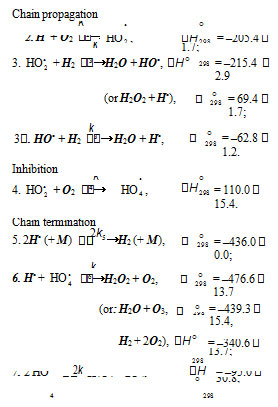
The chain evolution (propagation and inhibition) stage of the Scheme includes consecutive reaction pairs 2–3 and 3–3; the parallel (competing) reaction pair 3–4; and consecutive–parallel reactions 2 and 4
The hydroperoxyl free radical HO•2 [23–26] resulting energy released the conversion of the О=О multiple bond into the НО–О• ordinary bond. Therefore, before its possible decomposition, it can interact with a hydrogen or oxygen molecule as the third body via parallel (competing) reactions 3 and 4, respectively. The hydroxyl radical НО• that appears and disappears in consecutivefrom reaction 2 possesses an increased energy due to the
parallel reactions 3 (first variant) and 3 possesses additional energy owing to the exothermicity of the first variant of reaction 3, whose heat is distributed between the two products. As a consequence, this radical has a sufficiently high reactivity not to accumulate in the system during these reactions, whose rates are equal (V3 =V3) under quasi-steady-state conditions, according to the above scheme. Parallel reactions 3 (second, parenthesized variant) and 3 regenerate hydrogen atoms. It is assumed [9, 10] that the hydrotetraoxyl radical HO •4(first reported in [27–29]) resulting from endothermic reaction 4, which is responsible for the peak in the experimental rate curve (Fig. 2, curve 3), is closed into a five-membered [ОО─Н···ОО]• cycle due to weak intramolecular hydrogen bonding [30, 31]. This structure imparts additional stability to this radical and makes it least can stem from the fact that its mean lifetime in water at 294 K, which is (3.6 ± 0.4) × 10–5 s (as estimated [11] from the value of 1/k for the monomolecular decay
reaction HO •⎯⎯k →HO •+O2 [32]), is 3.9 times longer than that of the linear HO3 radical [16, 33] estimated
the same way [11] for the same conditions [34], (9.1 ± 0.9) × 10–6 s.in MP2/6-311++G** calculations using the Gaussian-98
program confirmed that the cyclic structure of HO •4[35] is energetically more favorable than the helical structure [16] (the difference in energy is 4.8–7.3 kJ mol–1 depending on the computational method and the basis set).2 For example, with the MP2(full)/6-31G(d) method, the difference between the full energies of the cyclic and acyclic HO•4 conformers with their zero-point energie (ZPE) values taken into account (which reduces the energy difference by 1.1 kJ mol –1) is –5.1 kJ mol–1 and the entropy of the acyclic-to-cyclico HO •4 transition is S298 = −1.6 kJ mol–1 K–1. Therefore, under standard conditions,HO •4 can exist in both forms, but the cyclic structure is obviously dominant (87%, Keq = 6.5) [35]. Reaction 4 and, to a much lesser degree, reaction 6 inhibit the chain process, because they lead to inefficient consumption of its main participants – HO • and Н•.
The hydrogen molecule that results from reaction 5 in the gas bulk possesses an excess energy, and, to acquire stability within the approximation used in this work, it should have time for deactivation via collision with a particle M capable of accepting the excess energy [37]. To simplify the form of the kinetic equations, it was assumed that the rate of the bimolecular deactivation of the molecule substantially exceeds the rate of its monomolecular decomposition, which is the reverse of reaction 5 [38].
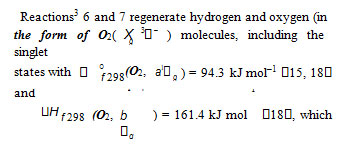
are deactivated by collisions, and in the form of O3) and yield hydrogen peroxide or water via a nonchain reactive.
mechanism,presumably through the intermediate formation of the unstable hydrogen tetraoxide molecule

2There were calculations for the two conformers (cis and trans) of the HO 4 radical [36] using large scale ab initio methods and density functional techniques with extended basis sets. Both conformers have a nearly planar geometry with respect to the four oxygen atoms and present an unusually long central O–O bond. The most stable conformer of HO4 radical is the cis one, which is computed to be endothermic with
Taking into account the principle of detailed balance for the various
pathways of formation of products, whose numbers in the elementary reaction should not exceed three for possible involvement in the triple collisions in the case of the reverse reaction, since the probability of simultaneous interaction of four particles is negligible.
The reaction of ozone with Н• atoms, which is not impossible, results in their replacement with НО• radicals. The relative contributions from reactions 6 and 7 to the process kinetics can be roughly estimated from the corresponding enthalpy increments (Scheme).H2O4 [39, 40].4 Ozone does not interact with molecular hydrogen. At moderate temperatures, it decomposes fairly slowly, particularly in the presence of О2( Χ 3− ) 18.
When there is no excess hydrogen in the hydrogen– oxygen system, the homomolecular dimer O4 [19–22, 41, 42], which exists at low concentrations (depending on the pressure and temperature) in equilibrium with O2 [18], can directly capture the Н• atom to yield the heteronuclear cluster5 HO • . This HO • cluster is more stable than O4 [18] and cannot abstract a hydrogen atom from the hydrogen molecule. Therefore, in this case, nonchain hydrogen oxidation will occur to give molecular oxidation products via the disproportionation of free radicals.
The low-reactive hydrotetraoxyl radicalHO •4 [32], which presumably has a high energy density [19], may be an intermediate in the efficient absorption and conversion of biologically hazardous UV radiation energy the Earth upper atmosphere. The potential energy surface for the atmospheric reaction HO• + О3, in which the adduct HO4 (2А) was considered as an intermediate, was calculated by the DMBE method [43]. From this standpoint, the following reactions are possible in the upper troposphere, as well as in the lower and middle stratosphere, where most of the ozone layer is situated (altitude of 16–30 km, temperature of 217–227 K, pressure of 1.0 104 – 1.2 103 Pa [44]; the corresponding kJ mol–1 [15]): 298 reaction values are given in

Staehelin et al. [32] pointed out that, in natural systems in which the concentrations of intermediates are often very low, kinetic chains in chain reactions can be very long in the absence of scavengers since the rates of the chain termination reactions decrease with decreasing concentrations of the intermediates according to a quadratic law, whereas the rates of the chain propagation reactions decrease according to a linear law.
The kinetic description of the noncatalytic oxidation of hydrogen, including in an inert medium [37], in terms of the simplified scheme of free-radical nonbranched-chain reactions (Scheme), which considers only quadratic-law chain termination and ignores the surface effects [5], at moderate temperatures and pressures, in the absence of transitions to unsteady-state critical regimes, and at a substantial excess of the hydrogen concentration over the oxygen concentration was obtained by means of quasi-steady-state treatment, as in the previous studies on the kinetics of the branched-chain free-radical oxidation of hydrogen [46], even though the applicability of this method in the latter case under unsteady states conditions was insufficiently substantiated. The method was used with the following condition6 for the first stages of the

The kinetic equations were derived for the rates (mol dm–3 s–1) of the elementary reactions for the formation of molecular products of hydrogen oxidation.
The rate of the chain formation of hydrogen peroxide in propagation reaction 3 and of water in reactions 3 and 3′ with V3, 3′(H2O) = 2V3 is

calculated using quantum chemical methods (B3LYP density functional theory) [40]. The hydrogen bond energy is 47.7 and 49.4 kJ mol–1 at 298 K for the triplet and singlet states of the dimer, respectively.
5It is impossible to make a sharp distinction between the two-step bimolecular interaction of three species via the equilibrium formation of the labile intermediate O4 and the elementary trimolecular reaction О2 +H→HO4.
6For example, the ratio of the rate constants of the bimolecular disproportionation and dimerization of free radicals at room temperature is k(HO• + HO2•)/2k(2HO•)2k(2HO2•)0.5 = 2.8 in the atmosphere [44] and k(H• + HO•)/2k(2H•)2k(2HO•)0.5 = 1.5 in water [3]. These values that are fairly close to unity.
7This equation can be used to describe a wide range of nonbranched- chain reactions of addition of any free radicals to the C=C bonds of unsaturated hydrocarbons, alcohols, etc., which result in the formation of molecular 1:1 adducts in binary reaction systems of saturated and unsaturated components [1, 9, 10]. x2 + (l + x)x2 l ; and 2k is the rate constant of
hydrogen atom recombination reaction 5 considered to be bimolecular in this approximation. The rate constant 2k5 in the case of the pulsed radiolysis of ammonia–oxygen (+ argon) gaseous mixtures at a total pressure of 105 Pa and a temperature of 349 K was calculated to be 1.6 × 108 dm3 mol–1 s–1 [6] (a similar value of this constant for the gas phase was reported in an earlier publication [48]). Pagsberg et al. [6] found that the dependence of the yield of the intermediate НО• on the oxygen concentration has a maximum close to 5 × 10–4 mol dm–3. In the computer simulation of the process, they
considered the strongly exothermic reaction HO •+ NН3→ Н2О + •NНОН, which is similar to reaction 3 in Scheme, whereas the competing reaction 4 was not taken modified Eqs. (3) and (4), in witch (αl + x) replaced with into account.The ratio of the rates of the competing reactions is V3/V4 = αl/x and the chain length is ν = V3/V1. Equation (1a) was obtained by substitution of the rate constant k2 into Eq. (1) with its analytical expression (in order to reduce the number of unknown parameters that are to be measured directly). The optimum concentration xm of oxygen, at which the rate of oxidation is maximum, can be calculated from Eq. (1a) or the analytical expression for k2 if other parameters that appear in these expressions are known.
The rates of nonchain formation of molecular hydrogen, hydrogen peroxide, and water in reactions 5, 6, and 7 of quadratic chain termination are as follows:

![]()

radical, rather than HO 4 , takes part in reactions 6 and 7. It is important to note that, if in the Scheme chain initiation via reaction 1 is due to the interaction between molecular hydrogen and molecular oxygen yielding the hydroxyl radical НО• instead of Н• atoms and if this radical reacts with an oxygen molecule (reaction 4) to form the hydrotrioxyl radical HO 3(which was obtained in the gas phase by neutralization reionization (NR) mass spectrometry [33] and has a lifetime of >10–6 s at 298 K) and chain termination takes place via reactions 5–7 involving the НО• and HO• , radicals instead of Н• and HO • , respectively, the expressions for the water chain formation rates derived in the same way will appear as a rational function of the oxygen concentration x without a

10–4 mol dm–3) at 296 K [7]. These data were calculated in the present work from the initial slopes of hydrogen peroxide buildup versus dose curves for a 60Co -radiation dose rate of Р = 0.67 Gy s–1 and absorbed doses of D 22.5–304.0 Gy. The following values of the primary radiation-chemical yield G (species per 100 eV of energy absorbed) for water -radiolysis products in the bulk of solution at pH 4–9 and room temperature were used
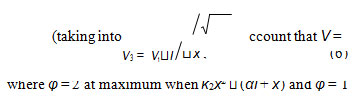
at the decreasing part of the curve. In the case of nonchain hydrogen oxidation via the GP and V1 = GHP): G 0.75 and GH = 0.6 (initiation yield) [3]; V1 = 4.15 10–8 mol dm–3 s–1 ; 2k5 = 2.0 × 1010 dm3 mol–1 s–1 3. As be seen from Fig. 1, the best description of the data with an increase in the oxygen concentration in water is attained when the rate V7 of the formation of hydrogen peroxide via the nonchain mechanism in the chain termination reaction 7 (curve 1, α = (8.5 2) 10–2) is taken into account in addition to the rate V3 of the chain formation of this product via the propagation reaction 3 (dashed curve 2, α = 0.11 0.026). The rate constant of addition reaction 2 determined from α is substantially underestimated: k2 = 1.34 107 (vs. 2.0 1010 [3]) dm3 mol–1 s–1. The difference can be due to the fact that the radiation-chemical specifics of the process were not considered in the kinetic description of the experimental data. These include oxygen consumption via reactions that are not involved in the hydrogen oxidation Scheme and reverse reactions resulting in the decomposition of hydrogen peroxide by intermediate products of water radiolysis ( e−, Н•, НО•), with the major role played by the hydrated electron [3].
Conclusion
Thus, the addition reaction of the HO • radical that possesses an elevated energy in statu nascendi with the oxygen molecule (at sufficiently high oxygen concentrations) to give the HO •radical was used for the
first time in the kinetic description of the initiated
hydrogen oxidation at moderate temperature and pressure [9, 10]. This reaction is endothermic and competes with radical generated in the former reaction has a low reactivity and inhibits the chain reaction.8the chain propagation reaction via the Н• atom. The HO • The above data concerning the competition kinetics of the nonbranched-chain addition of hydrogen atoms to the multiple bonds of the oxygen molecules make it possible to describe, using rate equations (1a) and (4a), obtained by quasi-steady-state treatment, the peaking experimental dependences of the formation rates of molecular 1:1 adduct H2O2 on the concentration of the oxygen over the entire range of its variation in binary system (Fig. 1). In such reaction systems consisting of saturated and unsaturated components [51, 52], the unsaturated compound (in this case the O2) is both a reactant and an autoinhibitor, specifically, a source of low-reactive free chains.radicals (in this case the HO • radicals) shortening kinetic The progressive inhibition of the nonbranched-chain processes, which takes place as the concentration of the unsaturated compound is raised (after the maximum process rate is reached), can be an element of the self- regulation of the natural processes that returns them to the stable steady state.
Using mechanism of the nonbranched-chain free-radical hydrogen oxidation considered here, it has been demonstrated that, in the Earth’s upper atmosphere, the decomposition of O3 in its reaction with the НО• radical can occur via the addition of the latter to the ozone molecule, yielding the HO 4 radical, which is capable of efficiently absorbing UV radiation [32].
References
- M. M. Silaev and L. T. Bugaenko, “Mathematical Simulation of the Kinetics of Radiation Induced Hydroxyalkylation of Aliphatic Saturated Alcohols”, Radiation Physics and Chemistry. 1992;40;1,1–10.
- M. M. Silaev, “Applied Aspects of the γ-Radiolysis of C1–C4 Alcohols and Binary Mixtures on Their Basis”, Khimiya Vysokikh Energii, Vol. 36, No. 2, 2002, pp. 97–101, English Translation in: High Energy Chemistry, 2002;36(2);70–74.
- A.K. Pikaev, “Sovremennaya radiatsionnaya khimiya. Radioliz gazov i zhidkostei” (“Modern Radiation Chemistry: Radiolysis of Gases and Liquids”), Nauka, Moscow. 1986.
- H. A. Smith and A. Napravnik, “PhotochemicalOxidation of Hydrogen”, Journal of the American Chemistry Society. 1940;62(1);385–393.
- E. J. Badin, “The Reaction between Atomic Hydrogen and Molecular Oxygen at Low Pressures. Surface Effects”, Journal of the American Chemistry Society, 1948;70(11);3651–3655.
- P. B. Pagsberg, J. Eriksen, and H. C. Christensen, “Pulse Radiolysis of Gaseous Ammonia–Oxygen Mixtures”, The Journal of Physical Chemistry. 1979;83(5);582–590.
- N. F. Barr and A. O. Allen, “Hydrogen Atoms in the Radiolysis of Water”, The Journal of Physical Chemistry. 1959;63(6);928–931.
- M. M. Silaev, “A New Competitive Kinetic Model of 03–209, EnglishTranslation in: Biophysics. 2001;46(2);202–207.
- M. M. Silaev, “The Competition Kinetics of Nonbranched Chain Processes of Free-Radical Addition to Double Bonds of Molecules with the Formation of 1:1 Adducts and the Inhibition by the Substrate”, Oxidation Communication. 1999;22(2);159–170.
- M. M. Silaev, “The Competition Kinetics of Radical– Chain Addition”, Zhurnal Fizicheskoi Khimii, Vol. 73, No. 7, 1999, pp. 1180–1184, English Translation in: Russian Journal of Physical Chemistry. 1999;73(7);1050–1054.
- M. M. Silaev, “Competitive Mechanism of the form of formaldehyde in nonmethanolic alcohol–formaldehyde systems [1, 9, 10] is that in the former does not include the formation of the molecular 1:1 adduct via reaction 4. Nonbranched Radical Chain Oxidation of Hydrogen Involving the Free Cyclohydrotetraoxyl Radical [ОО···Н···ОО]•, which Inhibits the Chain Process”, “Khimiya Vysokikh Energii, 2003, vol. 37, no. 1, pp. 27– 32, English Translation in: High Energy Chemistry. 2003;37(1);24–28.
- M. M. Silaev, “Simulation of the Initiated Addition of Hydrocarbon Free Radicals and Hydrogen Atoms to Oxygen via a Nonbranched Chain Mechanism”, Teoreticheskie Osnovy Khimicheskoi Tekhnologii, 2007, vol. 41, no. 6, pp. 634–642, English Translation in: Theoretical Foundation of Chemical Engineering.2007;41(6);831–838.
- M. M. Silaev, “Simulation of Initiated Nonbranched Chain Oxidation of Hydrogen: Oxygen as an Autoinhibitor”, Khimiya Vysokich Energii, 2008, vol. 42, no. 2, pp. 124–129, English Translation in: High Energy Chemistry.2008;42(1):95–100.
- N. N. Semenov, “Tsepnye reaktsii” (“Chain Reactions”), Goskhimtekhizdat, Leningrad. 1934;241, 203.
- J. S. Francisco and I. H. Williams, “The Thermochemistry of Polyoxides and Polyoxy Radicals”, International Journal of Chemical Kinetics. 1988;20(6):455–466.
- D. J. McKay and J. S. Wright, “How Long Can You Make an Oxygen Chain?”, Journal of the American Chemistry Society. 1998;120(5):1003–1013.
- N. P. Lipikhin, “Dimers, Clusters, and Cluster Ions of Oxygen in the Gas Phase”, Uspekhi Khimii. 1975;44(8):366–376.
- S. D. Razumovskii, “Kislorod – elementarnye formy i svoistva” (“Oxygen: Elementary Forms and Properties”), Khimiya, Moscow. 1979.
- K. M. Dunn, G. E. Scuceria, and H. F. Schaefer III, “The infrared spectrum of cyclotetraoxygen, O4: a theoretical investigation employing the single and double excitation coupled cluster method”, The Journal of Chemical Physics. 1990;92(10):6077–6080.
- A. Brown and V. Vaida, “Photoreactivity of Oxygen Dimers in the Ultraviolet”, The Journal of Physical Chemistry. 1996;100(19):7849–7853.
- V. Aquilanti, D. Ascenzi, M. Bartolomei, D. Cappelletti, S. Cavalli, M. de Castro-Vitores, and F. Pirani, “Molecular Beam Scattering of Aligned Oxygen Molecules. The Nature of the Bond in the O2–O2 Dimer”, Journal of the American Chemistry Society, 1999;121;(46):10794–1080.
- F. Cacace, G. de Petris, and A. Troiani, “Experimental Detection of Tetraoxygen”, Angewandte Chemie, Internation Edition (in English).2001;40;(21):4062–4065.
- H. S. Taylor, “Photosensitisation and the Mechanism of Chemical Reactions”, Transactions of the Faraday Society, 1926, vol. 21, no. 63(3), pp. 560–568.
- F. Cacace, G. de Petris, and A. Troiani, “Experimental Detection of Tetraoxygen”, Angewandte Chemie, Internation Edition (in English), 2001, vol. 40, no. 21, pp. 4062–4065.
- H. S. Taylor, “Photosensitisation and the Mechanism of Chemical Reactions”, Transactions of the Faraday Society, 1926, vol. 21, no. 63(3), pp. 560–568.
- S. N. Foner and R. L. Hudson, “Mass spectrometry of the HO2 free radical”, The Journal of Chemical Physics. 1962;36(10):2681.
- S. N. Foner and R. L. Hudson, “Mass spectrometry of the HO2 free radical”, The Journal of Chemical Physics.1962;36(10):2681.
- P. D. Lightfoot, B. Veyret, and R. Lesclaux, “Flash Photolysis Study of the CH3O2 + HO2 Reaction between 248 and 573 K”, The Journal of Physical Chemistry. 1990;94;(2):708–714.
- P. Smith, “The HO3 and HO4 Free Radicals”,Chemistry and Industry. 1954;(42):1299–1300.
- A. J. B. Robertson, “A Mass Spectral Search for H2O4 and HO4 in a Gaseous Mixture Containing HO2 and O2”, Chemistry and Industry. 1954;48:1485.
- D. Bahnemann and E. J. Hart, “Rate Constants of the Reaction of the Hydrated Electron and Hydroxyl Radical with Ozone in Aqueous Solution”, The Journal of Physical Chemistry. 1982;86(2):252–255.
- G. C. Pimentel and A. L. McClellan, “The Hydrogen Bond”, L. Pauling, Editor, Freeman, San Francisco, 1960;200.
- “Vodorodnaya svyaz’: Sbornik statei” (“The Hydrogen Bonding: Collection of Articles”) N. D. Sokolov, Editor, Nauka, Moscow. 1981.
- J. Staehelin, R. E. Bühler, and J. Hoigné, “Ozone Decomposition in Water Studied by Pulse Radiolysis. 2. OH and HO4 as Chain Intermediates”, The Journal of Physical Chemistry, 1984;88(24):5999–6004.
- F. Cacace, G. de Petris, F. Pepi, and A. Troiani, “Experimental Detection of Hydrogen Trioxide”, Science. 1999;285;5424:81–82.
- R. F. Bühler, J. Staehelin, and J. Hoigné, “Ozone Decomposition in Water Studied by Pulse Radiolysis. 1.HO2/O –and HO3/O3–as Intermediates”, The Journal of Physical Chemistry. 1984;88(12):2560–2564.
- I. V. Trushkov, M. M. Silaev, and N. D. Chuvylkin, “Acyclic and Cyclic Forms of the Radicals CH3O4 , and C2H5O4 : Ab Initio Quantum Chemical Calculations”, Izvestiya Akademii Nauk, Ser.: Khimiya, 2009, no. 3, pp. 479–482, English Translation in: Russian Chemical Bulletin, International Edition.2009;58(3):489–492.
- A. Mansergas, J. M. Anglada, S. Olivella, M. F. Ruiz-López, “On the Nature of the Unusually Long OO Bond in HO3 and HO4 Radicals”, Phys. Chem. Chem. Phys. 2007;9(44):5865–5873.
- W. Wong and D. D. Davis, “A Flash Photolysis Resonance Fluorescence Study of the Reactions of Atomic Hydrogen and Molecular Oxygen: H + O2 + M → HO2 + M”, International Journal of Chemical Kinetics. 1974;6;(3):401–416.
- S. W. Benson, “Thermochemical Kinetics: Methods for the Estimation of Thermochemical Data and Rate Parameters”, 2nd Edition, Wiley, New York, 1976.
- Yagodovskaya, T.V. and Nekrasov, L.I., “Use of Infrared Spectroscopy to Study Frozen Hydrogen– Oxygen Systems Containing Hydrogen Polyoxides”, Zhurnal Fizicheskoi Khimii. 1977;51(10):2434–2445.
- X. Xu. R. P. Muller, and W. A. Goddard III, “The Gas Phase Reaction of Singlet Dioxygen with Water: a Water-Catalyzed Mechanism”, Proceedings of the National Academy Sciences of the United States of America. 2002;99(6):3376–3381.
- E. T. Seidl and H. F. Schaefer III, “Is There a Transition State for the Unimolecular Dissociation of Cyclotetraoxygen (O4)?”, The Journal of Chemical Physics. 1992;96;(2):1176–1182.
- R. Hernández–Lamoneda and A. Ramírez–Solís, “Reactivity and Electronic States of O4 along Minimum Energy Paths”, The Journal of Chemical Physics. 2000;113;(10):4139–4145.
- A. J. C. Varandas and L. Zhang, “Test Studies on the Potential Energy Surface and Rate Constant for the OH + O3 Atmospheric Reaction”, Chemical Physics Letters. 2000;331(5–6):474–482.
- “Atmosfera. Spravochnik” (“Atmosphere: A Handbook”), Gidrometeoizdat, Leningrad. 1991.
- H. Okabe, “Photochemistry of Small Molecules”, Wiley, New York. 1978.
- A. B. Nalbandyan and V. V. Voevodskii, “Mekhanizm okisleniya i goreniya vodoroda” (“Mechanism of Hydrogen Oxidation and Combustion”), N. Kondrat’ev, Editor, Akad. Nauk SSSR, Moscow. 1949.
- L. Bateman, “Olefin Oxidation”, Quarterly Reviews. 1954;8(2):147–167.
- A. W. Boyd, C. Willis, and O. A. Miller, “A Re– examiation of the Yields in the High Dose Rate Radiolysis of Gaseous Ammonia”, Canadian Journal of Chemistry. 1971;49(13):2283-2289.
- M. M. Silaev, “Competition Mechanism of Substre– Inhibited Radical Chain Addition to Double Bond”, Neftekhimiya, 2000, vol. 40, no. 1, pp. 33–40, English Translation in: Petroleum Chemistry. 2000;40;(1):29–35.
- M. Silaev, Mathematical Simulation of Kinetics of Radiation-Induced Free-Radical Oxidation by Nonbranched-Chain Mechanism // Recent Innovations in Chemical Engineering. 2015;8(2):112–124.
- M. M. Silaev, “Low-reactive Free Radicals Inhibiting Nonbranched Chain Processes of Addition”, Biofizika, 2005, vol. 50, no. 4, pp. 585–600, English Translation in: Biophysics. 2005;50(4):511–524.
- M. M. Silaev, Kinetics of the Free-Radical Nonbranched-Chain Addition // American Journal of Polymer Science and Technology. 2017;3(3):29-49.