
Introduction
Prostate cancer is one of the main health problems worldwide [1,2]; it is important to mention that several drugs have been used for the treatment of this clinical pathology such as flutamide [3], apalutamide [4], bicalutamide [5], abiraterone [6], leuprolide [7], gosereline [8] and others; however, some of these drugs can cause several adverse effects. Therefore, in the search of other clinical alternative for treatment of prostate cancer have been synthesized several compounds; for exemple, the synthesis of an antiandrogen by addition of benzoazole to 3β-acetoxy-17-chloro-16-formylandrosta-5,16-diene [9]. Other study showed the preparation of a Cytochrome P450-17A1 antagonist via oximation of abiraterone as prostate cancer inhibitor [10]. In addition, a report indicates the preparation of a schiff base via reaction of Copper with quinoline-2 carboxaldehyde as proteasome inhibitors in human prostate Cancer Cells [11]. Other report showed the synthesis of an inhibitor of prostate cancer (17-Azolyl-steroid derivative) by the reaction of 3β-acetoxy-17-chloro-16-formylandrosta-5,16-diene with an azolyl group as nucleophile [12]. Also, a study showed the synthesis a prostate cancer inhibitor via acetylation of galeterone [13]. All these data indicate that several compounds can inhibit the prostate cancer; however, the site of interaction with some cell targets is not very clear, so more studies are needed on this phenomenon. Analyzing, this hypothesis, in this study three progesterone derivatives were synthesized, and a theoretical analysis was carried out on their interaction with androgen receptor or Cytochrome P450 17A1 enzyme using a docking model.
Experimental
Chemical synthesis
Progesterone bromide was prepared using a previously method reported [14]. In addition, all the reagents used in this study were purchased from Sigma-Aldrich Sigma-Aldrich Co., Ltd. The melting point for compounds was evaluated on an Electrothermal (900 model). Infrared spectra (IR) were determined using KBr pellets on a Perkin Elmer Lambda 40 spectrometer.1H and 13C NMR (nuclear magnetic resonance) spectra were recorded on a Varian VXR300/5 FT NMR spectrometer at 300 and 75.4 MHz (megahertz) in CDCl3 (deuterated chloroform) using TMS (tetramethylsilane) as an internal standard. EIMS (electron impact mass spectroscopy) spectra were determined using a Finnigan Trace Gas Chromatography Polaris Q-Spectrometer. Elementary analysis data were determined from a Perkin Elmer Ser. II CHNS/02400 elemental analyzer.
Preparation of 17-Acetyl-4-(6-hydroxy-hex-1-enyl)-10,13-dimethyl-1,2,6,7,8,9,10,11,12, 13,14,15,16,17-tetradecahydro-cyclopenta[a]phenanthren-3-one (2)
A solution of 4-bromideprogesterone (200 mg, 0.50 mmol), 5-hexyn-1-ol (70 µl; 0.63 mmol), Cooper(II) chloride anhydrous (67 mg, 0,5), and 5ml of methanol was stirring to reflux for 12 h. The mixture obtained was dried under reduced pressure and purified by crystallization using the methanol:water (4:1) system. yielding 54 %; IR (Vmax, cm-1) 3480, 3430, 3400, and 1722: 1H NMR (500 MHz, Chloroform-d) δH: O.68 (s, 3H), 1.08 (s, 3H), 1.16-1.56 (m, 8H), 1.58-1.62 (m, 4H), 1.70 (1H), 1.92 (broad, 1H), 1.94-2.10 (m, 5H), 2.12 (s, 3H), 2.19 (m, 1H), 2.28 (m, 2H), 2.32-2.54 (4H), 3.64 (m, 2H) ppm. 13C NMR (500 MHz, Chloroform-d) δC: 13.22, 18.44, 18.60, 20.78, 23.12, 23.72, 26.49, 27.22, 29.92, 31.87, 31.96, 34.24, 34.92, 35.80, 35.53, 36.60, 38.10, 43.82, 55.80, 56.00, 62.10, 63.40, 75.00, 114.20, 119.52, 165.50, 190.34, 206.62 ppm. EI-MS m/z: 410.28. Anal. Calcd. for C27H40O3: C, 78.98; H, 9.33; O, 11.69. Found: C, 78.90; H, 9.28.
Synthesis of of 6-[6-(1H-indol-2-yl)-1,5-dimethyl-16-azahexacyclo[11.11.0.0²,¹⁰.0⁵,⁹. 0¹⁵,²³.0¹⁷,²²] tetracosa-13,15(23),17(22),18,20-pentaen-14-yl]hex-5-yn-1-ol (3)
A solution of 2 (200 mg, 0.48 mmol), phenylhydrazine hydrochloride (100 mg; 0.69 mmol), and 8 ml of acetic acid:ethanol (3:5) was stirring to reflux for 8 h. The mixture obtained was dried under reduced pressure and purified by crystallization using the methanol:hexano:water (4:1:1) system. yielding 45 % of product; IR (Vmax, cm-1) 3480, 3430, and 2224: 1H NMR (500 MHz, Chloroform-d) δH: 0.78 (s, 3H), 1.00 (s, 3H), 1.16-1.50 (m, 6H), 1.56 (m, 2H), 1.57 (m, 1H), 1.62 (m, 2H), 1.70-2.10 (m, 8H), 2.20 (m, 2H), 2.34-2.44 (m, 2H), 3.66 (m, 2H), 3.70 (m, 1H), 6.00 (d, 1H, J = Hz), 6.80 (broad, 3H) and 6.86-7.60 (m, 8H) ppm. 13C NMR (500 MHz, Chloroform-d) δC: 13.80, 18.50, 19.54, 21.12, 23.39, 25.82, 26.47, 27.70, 31.82, 31.92, 35.32, 36.04, 36.72, 39.07, 39.19, 49.42, 52.22, 53.50, 62.04, 81.96, 98.25, 103.60, 106.12, 110.52, 112.04, 116.52, 118.62, 119.34, 119.82, 120.00, 120.38, 122.00, 127.24, 129.09, 133.68, 134.02, 134.80, 137.22 and 139.66 ppm. EI-MS m/z: 556.34. Anal. Calcd. for C39H44N2O: C, 84.13; H, 7.97; N, 5.03; O, 2.87. Found: C, 84.08; H, 7.90.
Preparation of 14-(6,7-dihydrooxepin-2-yl)-6-(1H-indol-2-yl)-1,5-dimethyl-16-azahexa- cyclo[11.11.0.0²,¹⁰.0⁵,⁹.0¹⁵,²³.0¹⁷,²²]tetracosa-13,15(23),17(22),18,20-pentaene (4)
A solution of 3 (200 mg, 0.36 mmol), phenylhydrazine hydrochloride (100 mg; 0.69 mmol), Cooper(II) chloride anhydrous (67 mg, 0,50 mmol), and 5ml of methanol was stirring to reflux for 8 h. The mixture obtained was dried under reduced pressure and purified by crystallization using the methanol:benzene:water (4:1:1) system. yielding 66 % of product, m.p. 118-120 oC; IR (Vmax, cm-1) 3430, 1625, and 1080: 1H NMR (500 MHz, Chloroform-d) δH: 0.80 (s, 3H), 1.02 (s, 3H), 1.04-1.48 (m, 6H), 1.50 (m, 1H), 1.58-1.76 (m, 3H), 1.77 (m, 1H), 1.78-2.40 (m, 8H), 3.50-3.58 (m, 2H), 3.70 (m, 1H), 5.60-5.96 (m, 2H), 6.00 (m, 1H), 6.26 (m, 1H), 6.86-7.70 (m, 8H), 9.20 (broad, 2H) ppm. 13C NMR (500 MHz, Chloroform-d) δC: 13.80, 18.45, 21.06, 25.83, 26.17, 27.73, 31.20, 34.39, 34.52, 35.80, 36.00, 39.06, 39.19, 49.44, 52.22, 53.62, 68.04, 103.63, 110.74, 111.32, 111.92, 112.08, 116.96, 118.76,119.31, 119.85, 120.03, 120.38, 122.00, 124.32, 125.62, 127.24, 127.22, 132.94, 134.00, 135.02, 137.22, 142.80 and 172.08 ppm. EI-MS m/z: 554.32. Anal. Calcd. for C39H42N2O: C, 84.44; H, 7.63; N, 5.05; O, 2.88. Found: C, 84.40; H, 7.58.
Electronic parameters evaluation (HOMO and LUMO)
The molecular orbitals HOMO and LUMO for all compounds were theoretically evaluated with SPARTAN’06 software package (Wavefunc-tion Inc. Irvine, CA, 2000), using Hartree-fock method at 321-G level [15].
Interaction between compounds 1-4 with androgen receptor and Cytochrome P450 17A1
Theoretical analysis of interaction of compounds 1–4 on androgen receptor (3L3Z) [16] and citocromo P450 17A1 enzyme (3RUK) [17] was carried out using a docking program (DockingServer) [18]. In addition, flutamide and abiraterone were used as controls.
Results and Discussion
There are several studies which indicate that some compounds can inhibit prostate cancer [9-13]; however, the site of interaction with some cell target is not very clear, so more studies are needed on this phenomenon. Analyzing this premise, the aim of this study was synthesized three progesterone derivatives to evaluate their interaction with androgen receptor or Cytochrome P450 17A1 enzyme.
First stage
Preparation of a steroid-propargyl-alcohol derivative (2)
There are several reports which showed the preparation of some propargylic-alcohols using different methods and reagents such as disulfide-oxazolidine10, Ti(O-i-Pr)4-BINOL complex [19], chiral diamine-coordinated tin(II) triflate [20], P(PhCH2NCH2CH2)3N [21] and others; however some of these reagents are difficult to handle require and special conditions. Therefore, in this work the estrone was reacted with 5-Hexyn-1-ol in basic medium (Figure 1). The mechanism of reaction involves a mechanism via SN2. The results of 1H NMR spectrum of 2 showed several signals at 0.68-1.08 ppm for methyl groups bound to steroid nucleus; at 2.12 ppm for methyl group bound to ketone; at 1.16-1.56, 1.70, 1.94-2.10, 2.19 and 2.32-2.54 ppm for steroid moiety; at 1.58-1.62, 2.28 and 3.64 ppm for methylene groups involved in the arm bound to alkene group; at 1.92 ppm for hydroxyl group. The 13C NMR spectra displays chemical shifts at 13.22-18.44 ppm for methyl groups bound to steroid nucleus; at 20.78-23.72, 27.22, 31.96-56.00, 63.40, 132.04 and 119.52-165.50 ppm for steroid moiety; at 18.60, 26.49, 31.87 and 62.10 ppm for methylene groups of arm which are bound to ring A of steroid; at 75.00-114.20 ppm for alkyne group; at 190.34-206.62 for ketone groups. In addition, the mass spectrum from 2 showed a molecular ion (m/z) at 410.28.
Synthesis of an indole-steroid-propargyl derivative (3)
It is important to mention that there are several procedures which are available for synthesis of indole derivatives; nevertheless, expensive reagents and special conditions are required [22-25]; in this study, 2 was reacted with phenylhydrazine under mild conditions (Figure 1). The results of 1H NMR spectrum of 3 showed several signals at 0.78-1.00 ppm for methyl groups bound to steroid nucleus; at 1.16-1.50, 1.57, 1.70-2.10, 2.34-2.44 and 3.70 ppm for steroid moiety; at 1.56, 1.62, 2.20 and 3.66 ppm for arm bound to ring A of steroid; at 6.00-7.60 ppm for both indole groups; at 6.80 for both amino and hydroxyl groups. The 13C NMR spectra displays chemical shifts at 13.80 and 19.54 ppm for methyl groups bound to steroid nucleus; at 21.12-25.82, 27.70, 31.92-53.50, 106.12 and 139.66 ppm for steroid moiety; at 18.50, 26.47, 31.82 and 62.04 ppm for arm bound to ring A of steroid; at 81.96-98.25 ppm for alkyne group; at 103.60 and 110.52-137.22 ppm for both indole groups. Finally, the mass spectrum from 3 showed a molecular ion (m/z) at 556.34.
 |
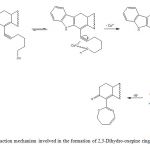 |
Figure 2: Reaction mechanism involved in the formation of 2,3-Dihydro-oxepine ring. |
Formation of 2,3-dihydroxepine ring
There are several reagents for preparation of oxepine derivatives such as terminal alkynes [26], senecialdehyde [27], palladium [28] and others. In this study, the compound 4 was synthesized via a intramolecular reaction (Figure 1 and 2) of 3 with Copper(II). The results of 1H NMR spectrum of 4 showed several signals at 0.80-1.02 for both methyl groups bound to steroid nucleus; at 1.04-1.48, 1.58-1.76, 1.76-2.40 and 3.70 ppm for steroid moiety; at 1.50, 1.77, 3.50-3.58, 5.60-5.96 and 6.26 ppm for 2,3-dihydroxepine ring; at 6.00, 6.86-7.70 ppm for both indole groups; at 9.20 ppm for amino groups. The 13C NMR spectra displays chemical shifts at 13.80-18.45 ppm for methyl groups bound to steroid nucleus; at 21.06-31.20, 34.52-53.62, 111.92, 129.62 and 142.80 ppm for steroid moiety; at 34.39, 68.04, 111.32, 124.32, 135.02 and 172.08 ppm for 2,3-dihydroxepine ring; at 103.63- 110.74, 112.08-122.03, 127.22-134.00 and 137.22 for both indole groups. Additionally, the mass spectrum from 4 showed a molecular ion (m/z) at 554.32.
 |
 |
Physicochemical parameters of compounds 1-4.
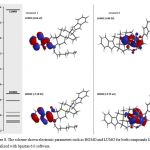
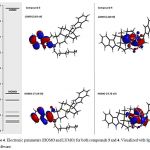
It is important to mention that some chemical characteristic of compounds 1-4 could condition their biological activity on some biological target; therefore, to evaluate this hypothesis some chemistry descriptors such as HOMO and LUMO were evaluated. The results (Figure 3-4 and table 1) indicates that HOMO was higher for the compound 4 compared with 1-3; these results indicate that 4 have a strong electro donating ability in comparison with 1-3 which could result in higher activity on some biological system compared with happening with another type of molecules [29].
|
|

 |
Table 1: Physicochemical parameters involved in the chemical structure of compounds 1-4.
| Parameter | Compound 1 | Compound 2 | Compound 3 | Compound 4 |
| Rotatable Bonds | 4 | 8 | 7 | 4 |
| cLogP | 5.446 | 5.339 | 9.231 | 9.846 |
| TPSA | 34.14 | 54.370 | 51.810 | 40.810 |
| HOMO | -10.30 | -9.32 | -7.19 | -5.75 |
| LUMO | 3.72 | 4.31 | 3.63 | 1.86 |
| Energy gap (HOMO-LUMO) | -14.02 | -13.63 | -10.82 | -7.61 |
| HBA | 2 | 3 | 1 | 1 |
| HBD | 0 | 1 | 3 | 2 |
Analyzing these data and other studies on structure-activity which suggest that other physiochemical factors involves of several drugs such as hydrogen bond donor groups (HBD) and hydrogen bond acceptor groups (HBA) may exert changes on some biological system [30]. In this regard, these physicochemical descriptors have been evaluated using some pharmacophore models [31-34]; It is important to mention that pharmacophores are generally used to evaluate the relationship between the structure and activity of a set of molecules; therefore, in this study a theoretical study was carried out using a pharmacophore model. [35]. The theoretical results (Figures 5 and 6; Table 1) showed several hydrogen bond donor groups (-OH) for the compounds 2; -NH- for 3 and 4. Other theoretical data showed several hydrogen bond acceptor groups such as -C=O for 1 and 2. In addition, other theoretical results (table 1) show both HBA and for HBD (< 5) values for compounds 1 to 4. Analyzing these results and other reports about Lipinski’s rule [36] which indicates that both HBD and HBA can condition some pharmacokinetic process of drugs in the human body; therefore, theoretical data suggest that compounds 2 to 7 could have the ability of penetrate some barrier biological of human body.
Table 2: Residues aminoacids involved in the interaction between flutamide and compounds 1-4 on androgen receptor (3L3Z). Similar aminoacid residues to flutamide (red).
| Flutamide | Compound1 | Compound2 | Compound3 | Compound4 | |
| Amioacid Residues | Leu704
Asn705 Trp741 Met742 Met745 Val746 Met749 Phe764 Met780 Leu873 Phe876 Thr877
|
Leu701
Leu704 Asn705 Leu707 Gln711 Trp741 Met742 Met745 Val746 Met749 Phe764 Met780 Leu873 Thr877 Leu880
|
Leu701
Leu704 Asn705 Leu707 Gln711 Trp741 Met742 Met745 Ala748 Met749 Arg752 Phe764 Met780 Phe876 Thr877 Leu880 Phe891 Met895 |
Leu701
Leu704 Asn705 Leu707 Trp741 Met742 Met745 Val746 Met749 Leu762 Phe764 Phe770 Ser778 Met780 Cys784 Met787 Leu873 Phe876 Thr877 Leu880 Phe891 Met895 |
Leu701
Leu704 Asn705 Gln711 Trp741 Met742 Met745 Val746 Met749 Phe764 Met780 Gln783 Met787 Leu873 Phe876 Thr877 Leu880 Phe891 Met895 |
Theoretical analysis of interaction of compounds 1-4 with androgen receptor (3L3Z) and citocromo P450 17A1 enzyme (3RUK).
Interactions between molecule-protein and protein-protein are involved in several biological processes such as signal transduction, physiological regulation, gene transcription, and enzymatic reactions [37]. It is important to mention that several drugs can induce changes biological activity of some biological system via interactions with either specific protein or enzyme; therefore, several theoretical models have been developed to predict the interaction of drugs with different proteins or enzymes [38]. Analyzing these data, in this study was carried out a theoretical analysis on interaction of compounds 1-4 with androgen receptor (3L3Z) or citocromo P450 17A1 enzyme (3RUK) using flutamide and abiraterone as control. The results showed (Table 2 and 3) the interaction of compounds 1-4 with several amino acid residues involved 3L3Z protein. These data indicate that flutamide could interact with several aminoacid residues such as Leu704, Asn705, Trp741, Met742, Met745, Val746, Met749, Phe764, Met780, Leu873, Phe876, Thr877 which are involved in the 3L3Z protein surface. It is important to mention, that also the compound 3 and 4 could bound to these aminoacid residues; however, only some of these aminoacid residues may participate in the interaction between 3L3Z protein with compounds 1 and 2.
Table 3: Residues aminoacids involved in the interaction between abiraterone e and compounds 1-4 on citocromo P450 17A1 enzyme (3RUK). Similar aminoacid residues to abiraterone (red).
| Abiraterone | Compound1 | Compound2 | Compound3 | Compound4 | |
| Amioacid Residues | Ala113
Tyr201 Asn202 Ile205 Asp298 Ala302 Thr306 Val366 Ala367 Val482
|
Ala113
Phe114 Asn202 Ile206 Leu209 Ala302 Glu305 Thr306 Val366 Ala367 Ile371 Val482
|
Ala113
Phe114 Tyr201 Asn202 Ile205 Ile206 Leu209 Arg239 Asp298 Ala302 Glu305 Thr306 Val366 Ala367 Ile371 Val482
|
Ala105
Ser106 Ala113 Phe114 Ile198 Asn202 Ile205 Ile206 Ile206 Leu209 Arg239 Leu243 Thr294 Asp298 Phe300 Glu305 Ala367 Ile371 Val482
|
Ser106
Ala113 Phe114 Ile198 Tyr201 Asn202 Ile205 Ile206 Ile206 Leu209 Arg239 Leu243 Thr294 Asp298 Glu305 Ile371 Val482
|
On the other hand, other results showed that abiraterone and compound 2 could interaction with same aminoacid residues of 3RUK protein surface such as Ala113, Tyr201, Asn202, Ile205, Asp298, Ala302, Thr306, Val366, Ala367, Val482; however, the compounds 1, 3 and 4 only interaction with some these types of aminoacid residues; this phenomenon may due to differences in the chemical structure of compounds. However, to validate this premise, other types of factors must be evaluated, such as some thermodynamic parameters.
Table 4: Thermodynamic parameters involved in the interaction of flutamide and compounds 1-4 on androgen receptor (3L3Z).
| Compound | Est. Free Energy of Binding (kcal/mol) | vdW + Hbond + desolv. Energy (kcal/mol) | Electrostatic
Energy (kcal/mol) |
Total Intermol. Energy (kcal/mol) | Interact. Surface |
| Flutamide | -7.40 | -8.48 | -0.02 | -8.50 | 441.685 |
| 1 | -10.26 | -10.55 | -0.01 | -10.56 | 536.496 |
| 2 | 8.00 | 6.19 | -0.15 | 6.04 | 688.692 |
| 3 | 259.02 | 258.28 | 0.01 | 258.29 | 888.947 |
| 4 | 233.04 | 234.41 | 0.12 | 234.53 | 850.152 |
Table 5: Thermodynamic parameters involved in the interaction of arbiterone and compounds 1-4 on Citocromo P450 17A1 enzyme (3RUK).
| Compound | Est. Free Energy of Binding (kcal/mol) | vdW + Hbond + desolv. Energy (kcal/mol) | Electrostatic
Energy (kcal/mol) |
Total Intermol. Energy (kcal/mol) | Interact. Surface |
| Arbiraterone | -11.43 | -12.05 | -0.01 | -12.05 | 596.198 |
| 1 | -10-10 | -10.43 | 0.04 | -10.39 | 536.563 |
| 2 | -9.87 | -11.77 | -0.07 | -11.83 | 677.752 |
| 3 | 13.07 | 11.66 | 0.06 | 11.72 | 903.028 |
| 4 | 18.04 | 19.16 | -0.04 | 19.12 | 849.271 |
Thermodynamic parameters
There are some reports which indicate that several thermodynamic factors may be involved in the interaction drug-protein [39]; therefore, in this study a theoretical ass was carried out on some thermodynamic parameters involved in the interaction of compounds 1-4 with androgen receptor (3L3Z) or Cytochrome P450 17A1 enzyme (3RUK) using flutamide or abiraterone as control. It is important to mention that thermodynamic parameters were following; 1) free energy of binding which determinate the energy value that require a molecule to interact with a protein in a water environment. 2) Electrostatic energy that is the product of electrical charge and electrostatic potential, which are involved in the ligand-protein system [40]; 3) total intermolecular energy and 4) Van der Waals (vdW) + hydrogen bond (Hbond) + desolvation energy (which have an influence on the movement of water molecules into or out of the ligand-protein system). The results showed in the table 4 indicate that energetic requirements involved in the interaction of progesterone derivatives with 3L3Z protein were higher for compound 3 compared with flutamide, compounds 1-2 and 4. However, other results (Table 5) showed that energy produce by interaction of 4 with 3RUK protein was higher in comparison with compounds 1-3. These data suggested that differences in the energy levels between the interaction of the compounds studied with the both 3L3Z and 3RUK proteins can be translated as different changes in the biological activity of aromatase in the presence of 3 or 4 in comparison with the compounds 1 and 2.
Conclusions
Theoretical results indicate that 1) the compounds 3 and 4 may act as androgen receptor inhibitors; 2) compound 2 could inhibit the biological activity of cytochrome P450 17A1 enzyme. All these data indicate that compounds 2, 3 and 4 could be a good candidates to prostate cancer.
References
- Collins, A.;Berry, P.; Hyde, C.; Michael, J.;Cancer Res. 2005, 65(23):10946-10951.
- Ortelli, L.; Spitale, A.; Mazzucchelli, L.; Bordoni, A. BMC Cancer. 2018, 18(1):733.
- Rezaeifar, Z.; Rounaghi, G.; Es’haghi, Z.; Chamsaz, M.; 2018, 91:10-18.
- Rachner, T.; Tsourdi, E.; Hofbauer, L.; Engl. J. Med. 2018, 378(26):2541-2542.
- Beebe, J.; Ruterbusch, J.; Bylsma, L.; Gillezeau, C.; Fryzek, J.; Schultz, N.; Flanders, S.; Barlev, A.; Ther. 2018, Jun 26. doi: 10.1007/s12325-018-0738-5.
- Bono, J.; Logothetis, C.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.; Jones, R.; Goodman, O.; Saad, F.; Staffurth, J.; Mainwaring, P.; Engl. J. Med. 2011, 364:1995-2005.
- Saltzstein, D.; Shore, N.; Moul, J.; Chu, F.; Concepcion, R.; Motte, S.; McLane, J.; Atkinson, S.; Yang, A.; Crawford, E.; T Adv. Urol. 2017, 10(2):43-50.
- Meani, D.; Solarić, M.; Visapää, H.; Rosén, R.; Janknegt, R.; Soče, M.; Adv. Urol. 2017, 10(2):51-63.
- Handratta,; Vasaitis, T.; Njar, V.; Gediya, L.; Kataria, R.; Chopra, P.; Newman, D.; Farquhar, R.; Guo, Z.; Qiu, Y.; Brodie, A.; J. Med. Chem. 2005, 48(8):2972-2984.
- Fehl, C.; Vogt, C.; Yadav, R.; Li, K.; Scott, E.; Aubé,; J. Med. Chem. 2018, 61(11): 4946-4960.
- Adsule,; Barve, V.; Chen, D.; Ahmed, F.; Dou, P.; Padhye, S.; Sakar, F.; J. Med. Chem. 2006, 49(24): 7242-7246.
- Njar,; Kato, K.; Nnane, I.; Grigoryev, D.; Long, B.; J. Med. Chem. 1998, 41(6); 902-912.
- Purushottamachar,; Godbole, A.; Gediya, L.; Martin, M.; Vasaitis, T.; Kwegyir, A.; Ramalingam, S.; Ates, Z.; Njar, V.; J. Med. Chem. 2013, 56(12):4880-4898.
- Allen, A.; Weiss M.; 16-alpha,17-alpha-isoalkylidenedioxy-4-pregnene-3,20-diones. USA. Patent. No. 3,001,991, Sept. 26, 1961.
- Hladka, I.; Lytvyn, R.; Volyniuk, D.; Gudeika, D.; Grazulevicius, J.; Dyes Pigm. 2018, 149:812-821.
- Issa, N.; Peters, O.; Byers, S.; Dakshanamurthy; Comb. Chem. High. Throughput. Screen. 2015, 18(8):784-794.
- Kuzikov, A.; Dugin,; Stulov, S.; Shcherbinin, D.; Steroids. 2014, 88:66-71.
- Mohamed,; Abdelhamid, A.; Almutairi, F.; Ali, A.; Bishr, M.; Bioorg. Med. Chem. 2018; 26(3):623-629.
- Marshall, J.; Bourbeau, M.; Lett. 2003, 5: 3197-3199.
- Mukaiyama, T.; Furuya, M.; Ohtsubo, A.; Kobayashi, S.; Lett. 1991, 20: 989992. [21] Wadhwa, K.; Chintareddy, V.; Verkade, J.; J. Org. Chem. 2009, 74:6681-6690.
- Fagnola, M.; Candiani, I.; Visentin, G.; Tetrahedron Lett. 1997, 38(13):2307-2310.
- Lee, D.; Cho, C.; Kim,J.; Youn, Y.; Shim, S.; Song, H.; Korean Chem. Soc. 1996, 17:1132-1135.
- Yasuhara, A.; Kanamori, Y.; Kancko, M.; Numata, A.; Kondo, Y.; Sakamoto, T.; Chem. Soc. Perkin Trans. 1999, 1(4):529-534.
- Haugland, R.; Yguerabide, J.; Stryer, L.; Nat. Acad. Sci. 1969, 63(1):23-30.
- Kuntala, N.; Telu, J.; Banothu, V.; Nallapati, S.; Anireddy, J.; Pal, S.; Chem. Commun. 2015,6:1612-1619.
- Paduraru, M.; Wilson, P.; Lett. 2003, 5(25):4911-4913.
- Shubhankar, S.; Hemakesh, M.; Rathin, J.; Jayanta, K.; Tetrahedron Lett.
- 2008, 49(50):7153-7156.
- Moreira, V.; Salvador, A.; Beja, A.; Paixao, J.; Steroids. 2011, 76(6):582-587.
- Moreira, V.; Vasaitis, T.; Njar, C.; Salvador, J.; Steroids. 2007, 72(14): 939-948.
- Figueroa-Valverde, L.; Rosas-Nexticapa, M.; Mateu-Armand V.; Herrera-Meza S.; Lopez-Ramos M.; García-Cervera E.; Pool-Gómez E.; Biointerfase Res. Chem. 2018, 8(3):3161-3169.
- Kerdawy, A.; Tautermann, C.; Clark, T.; Fox, T.; Chem. Inf. Model. 2013, 53(12): 3262-3272.
- Ranu, S.; Singh, A.; Chem. Inf. Model. 2011, 51(5):1106-1121.
- Koes, D; Camacho, C.; Chem. Inf. Model. 2011, 51(6):1307-1314.
- Wolber, G.; Langer, T.; Chem. Inf. Model. 2005, 45(1):160-169.
- Veber D, Johnson R., Yuan H.; Smith B.; ward K.; Med. Chem. 2002, 45(12):2615-2623.
- Andersson, D.; Thysell, E.; Lindström, A.; Bylesjö, M.; Raubacher, F.; Linusson, A.; Chem. Inf. Model. 2007, 47(4):1673-1687.
- Elokely, K.; Doerksen, R.; Chem. Inf. Model. 2013, 53(8):1934-1945.
- Dei,; Gatteschi, D.; Pardi, L.; Inorg. Chem. 1993, 32(8):1389-1395.
- Mandell, J.; Roberts, V.; Pique, M.; Kotlovyi, V.; Mitchell, J.; Nelson, E.; Tsigelny, I.; Ten, E.; Protein Engineering. 2001, 14(2): 105-113.